

Abstract
We report non-adiabatic molecular dynamic simulations of the ring opening reaction of diarylethene (DAE) derivative molecules, both free standing and embedded between gold electrodes. Simulations are performed by the surface hopping method employing density functional theory. Typically, the free-standing molecules exhibit large quantum yields to open and close; however the process is quenched for the molecules embedded between electrodes. Our simulations reveal the importance of the DAE side chemical groups, which explain the efficiency of the quenching process. Namely, delocalization of the LUMO state contributes to electronic coupling between the molecule and electrodes, suppressing or enhancing the reaction process. The simulations indicate that a proper choice of the chemical side group, which provides the strong localization of the LUMO state, can substantially diminish the quenching mechanism. Additionally, we analyze a strong dependency of the quantum yield of the opening reaction coming from the mechanical strength of the molecules.
Export citation and abstract BibTeX RIS
1. Introduction
One of the basic concepts of molecular electronic devices is molecular switches. Various molecules have been studied in solution as potential molecular switches [1]. However, to explore potential applications the molecular switches should also be performed on a gated molecule between metallic electrodes. It has been shown that the presence of metallic electrodes can significantly influence the functionality of the switching devices [2]. A deeper understanding of the metallic electrode effects on the switching mechanism of molecular contacts is desirable, and understanding gated molecules is one of the greatest challenges of molecular electronics [3].
A class of molecular switches based on diarylethene (DAE) molecules is a promising candidate for fabricating a molecular switch device [4]. The primary component of DAE-based molecules consists of the cyclohexadienyl (CHD) molecule, which is an active unit where the switching mechanism occurs. Illumination by ultraviolet or visible light initiates a transformation process of the molecule yielding an open to closed form, and vice versa. This cyclo-reversion reaction follows the Woodward–Hoffmann rules for symmetry-conserved conrotatory modes [5]. That is, the visible light excites an electron from the highest occupied molecular orbital (HOMO), having a bonding character, to the lowest unoccupied molecular orbital (LUMO) with an anti-bonding character. The electron excitation from HOMO to LUMO debilitates the σ bond between two carbon atoms in the switching CHD unit (see figure 2). Simultaneously the electron transfer reinforces the π bond of the C–C bond. Consequently, this effect initiates the opening process by breaking the C–C bond in the CHD part of the DAE-derivates. During the reaction the two frontier molecular orbitals HOMO and LUMO exchange their energetic positions, where the HOMO becomes LUMO and vice versa [6].
The details of the mechanism of the cyclo-reversion reaction in a CHD molecule is well understood [7]. Most of the studies regarding cyclo-reversion reactions of molecules containing the CHD ring describe potential energy profiles for static conformations along the reaction pathway [8–10]. In the last few years, a non-adiabatic molecular dynamic (NAMD) of such free standing molecules were also reported [11–16]. For example, the different dynamics for N- and I-type DAEs were simulated by surface hopping based on linear-response time dependent density functional theory (DFT) [13]. These simulations predicted the reaction quantum yields for N-type 55% and I-type 77% and reaction times 280 fs and 180 fs respectively [13]. Similar DAE derivatives were studied experimentally by ultrafast transient absorption spectroscopy which revealed quantum yields of only 7.5% and lifetimes of 1.3 ps [17].
The switching mechanism of free-standing molecules is well understood, but there is little known about how this process changes in molecular junctions where the molecule is gated to the metallic electrodes. The impact of the metallic electrodes on the switching process of a molecular junction was examined for the first time by Dulic et al [18]. They investigated experimentally the influence of gold electrodes on the switching properties of a DAE-derivative by means of a mechanically controllable break junction. The measurements showed that only a one-directional close-to-open switching mechanism is feasible. The switching in the opposite direction was not experimentally observed. They attributed the quenching of the first excited state of the open molecule to the presence of the gold electrodes [18]. Later, Meng et al fabricated a complex molecular junction made of a DAE switching unit and ruthenium acetilide arms gated between gold electrodes, which showed bidirectional switching characteristics under illumination [19]. They emphasized an importance of ruthenium acetilide arms on the switching process, which significantly decreases the electronic coupling of dithyenylethene fragments with the metal electrodes in its open form. This indicates that different side chains of the DAE molecule can have significant influence on the switching mechanism. The impact of the side chains of DAE-based molecules on their transport properties was also addressed by Kim et al [20].
In general, there are limited theoretical studies of the switching mechanism of molecular junctions, where a molecule is gated to metallic electrodes. Switching and transport properties of the DAE molecule modified by thiophene and benzene bridging gold electrodes were studied by using ground state total energy DFT simulations [2]. In the study, the analysis of the electronic structure and transport properties revealed strong electronic coupling of the LUMO orbital in the closed form of a molecule to the gold electrodes. This observation indicates suppression of the switching closed-to-open mechanism. Similar observations were made by Ashraf et al [21] investigating DAE derivative molecules bridging carbon nanotubes (CNT). Their theoretical analysis also revealed the inhibition of the closed-to-open photo-switching mechanism due to the strong coupling of the LUMO in the closed isomer to electronic states of CNT contacts. They estimated the lifetime of the excited state of the closed form to be about 7–10 fs from the transmission function obtained from ground state transport DFT calculations. Nevertheless, proper understanding of the quenching process due to the strong electron coupling of molecular orbitals to nuclear motion would require NAMD of excited states accounting for the entire system, including metallic electrodes. Such advanced simulations, to our knowledge, have been neglected thus far.
Here, we present NAMD simulations of the cyclo-reversion ring opening reaction of purely organic DAE derivatives, both single molecules and molecules embedded between the gold electrodes. We deliberately choose three types of the DAE derivates with different side groups (see figure 2), to analyze the impact of the electrodes and mechanical stress on the quenching process.
2. Simulation methodology
We utilized the NAMD implemented [22, 23] within the DFT Fireball code [24, 25] to simulate the cycloreversion reactions. The NAMD approach within Fireball is a local orbital implementation of the molecular dynamics with electronic transitions driven by the fewest switches surface hopping algorithm [26]. Simulations reported here were performed with an ionic time step of 0.5 fs and an electronic time step of 0.005 fs. The self-consistency criteria was  of the square of the charge orbital difference [27]. Atomic velocities were randomly generated from the temperature at the beginning of the simulation. The temperature was set to 300 K. We chose the following cut off radii of numerical atomic orbitals [28]: hydrogen,
of the square of the charge orbital difference [27]. Atomic velocities were randomly generated from the temperature at the beginning of the simulation. The temperature was set to 300 K. We chose the following cut off radii of numerical atomic orbitals [28]: hydrogen,  a.u., with an additional
a.u., with an additional  -state,
-state,  a.u.; carbon,
a.u.; carbon,  a.u,
a.u,  a.u., and d-state,
a.u., and d-state,  a.u.; sulphur,
a.u.; sulphur,  a.u.,
a.u.,  a.u.
a.u.  a.u.; nitrogen,
a.u.; nitrogen,  a.u.,
a.u.,  a.u.,
a.u.,  a.u.; and gold,
a.u.; and gold,  a.u.,
a.u.,  a.u.,
a.u.,  a.u. All simulations lasted 250 fs excluding DAE–butane (but); the simulation of DAE–but lasted 500 fs because of the longer reaction time required. Initial geometries of the ensemble of the trajectories were chosen randomly from geometries of the ground state Born–Oppenheimer molecular dynamics simulation. All NAMD simulations started from a single excited state, where one electron is excited from the HOMO to the LUMO, which mimics a promotion of an electron due to its interaction with light. The feasibility of this electronic transition was confirmed by calculation of the oscillator strength between the HOMO LUMO. Non adiabatic couplings were calculated in each time step using an analytical formulae [22] for the electronic states obtained from self-consistent Kohn–Sham orbitals corresponding to the given excited occupancy. In the case of the extended system, the electrodes were designed from a two layer slab containing 38 Au atoms. We did not apply periodic boundary conditions for the simulations and the electrodes were treated as a gold fixed cluster. We performed ground state molecular dynamics of a given system for 2000 time steps and then randomly choose 100 molecular configurations as initial geometries.
a.u. All simulations lasted 250 fs excluding DAE–butane (but); the simulation of DAE–but lasted 500 fs because of the longer reaction time required. Initial geometries of the ensemble of the trajectories were chosen randomly from geometries of the ground state Born–Oppenheimer molecular dynamics simulation. All NAMD simulations started from a single excited state, where one electron is excited from the HOMO to the LUMO, which mimics a promotion of an electron due to its interaction with light. The feasibility of this electronic transition was confirmed by calculation of the oscillator strength between the HOMO LUMO. Non adiabatic couplings were calculated in each time step using an analytical formulae [22] for the electronic states obtained from self-consistent Kohn–Sham orbitals corresponding to the given excited occupancy. In the case of the extended system, the electrodes were designed from a two layer slab containing 38 Au atoms. We did not apply periodic boundary conditions for the simulations and the electrodes were treated as a gold fixed cluster. We performed ground state molecular dynamics of a given system for 2000 time steps and then randomly choose 100 molecular configurations as initial geometries.
3. Non-adiabatic molecular dynamic simulations of free molecules
Figure 2 represents the atomic and electronic structure of free-standing DAE derivatives under study including two different side groups pyridine (pyr) and butane. The remarkable difference between the derivates is the spatial localization of the LUMO orbitals.
Namely, the LUMO orbital of the DAE–pyr is spread over the whole molecule including the pyridine side groups containing conjugation. Contrarily, in the case of DAE–but, the LUMO orbital is more localized on the switching unit which does not contain conjugation on the side groups. This difference does not influence the switching reaction of free-standing molecules, but it has significant influence when molecules are gated between electrodes, as will be discussed later.
First we analyzed the activation barriers for both the ground and the first excited states along the reaction pathway of the opening process, summarized in table 1. The pathway was obtained from nudge elastic band (NEB) [29] simulations. The excited barrier was calculated for the same pathway but for the excited state of the molecule. The calculated activation energy barriers of the excited state are much lower than those of the ground states, in good agreement with experimental evidence and previous theoretical calculations [13]. Also the presence of the side groups significantly decreases the activation barriers with respect to the basic DAE molecule. We also find that there is no correlation between the localization of the LUMO orbital and the activation barrier either in the ground state or in the excited state.
Table 1. Table of quantum yield of the reaction, barrier height Eb of the reaction calculated from the nudge elastic band simulations and reaction times treac of the ring opening reactions.
| Open/close | Eb [eV] GS/ES | treac [fs] | |
|---|---|---|---|
| DAE | 90/10 | 1.10/0.22 | 56.72 |
| DAE–pyr | 98/2 | 0.76/0.1 | 44.95 |
| DAE–but | 89/11 | 0.27/0.03 | 275.08/84.00(strait) |
Table 2. Table of quantum yield of the reaction, barrier height Eb of the reaction in the ground state calculated from the nudge elastic band simulations and reaction times treac of the ring opening reactions.
| Open/close | Eb [eV] | treac [fs] | |
|---|---|---|---|
| DAE Au | 0/100 | 0.5 | — |
| DAE–pyr Au | 0/100 | 1.3 | — |
| DAE–but Au | 58/42 | 1.2 | 261.41 |
After our initial barrier analysis, we performed NAMD simulations of free-standing molecules including 100 trajectories. The statistical analysis of the trajectory ensembles reveals high quantum yields  90% for the opening reaction in all three types of molecules (see table 1). Figure 3(A) displays the characteristic time evolution of the DAE molecule during the opening process. In particular, we track the evolution of the frontier orbital energies, their occupation and the characteristic bond length between two carbon atoms shown
90% for the opening reaction in all three types of molecules (see table 1). Figure 3(A) displays the characteristic time evolution of the DAE molecule during the opening process. In particular, we track the evolution of the frontier orbital energies, their occupation and the characteristic bond length between two carbon atoms shown  in figure 1. The potential energy surface together with the non-adiabatic coupling term [22] (gray line) are plotted in figure 3(B). After an initial electron excitation of an electron from HOMO to LUMO orbitals, the orbitals gradually approach each other in energy. We observe that the distance
in figure 1. The potential energy surface together with the non-adiabatic coupling term [22] (gray line) are plotted in figure 3(B). After an initial electron excitation of an electron from HOMO to LUMO orbitals, the orbitals gradually approach each other in energy. We observe that the distance  increases once the HOMO and LUMO orbitals approach each other by
increases once the HOMO and LUMO orbitals approach each other by  0.1 eV. After approximately 100 fs, the system approaches a conical intersection and the non-adiabatic coupling increases significantly, facilitating the transition.
0.1 eV. After approximately 100 fs, the system approaches a conical intersection and the non-adiabatic coupling increases significantly, facilitating the transition.
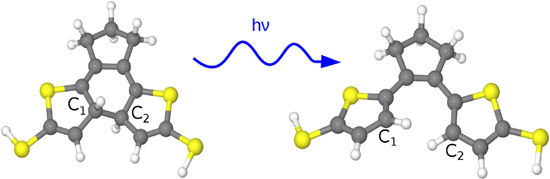
Figure 1. Atomic structure of a DAE molecule. The optimized molecule in the closed and open form is represented on the left and right respectively. The figure displays the atomic structure of the central ring including the definition of the C1–C2 bond.
Download figure:
Standard image High-resolution imageIn the proximity of the conical intersection, the single occupancy of HOMO and LUMO orbitals generate competing attractive and repulsive forces both acting on the C1–C2 bond. After  100 fs, the system evolves such that the energy of HOMO/LUMO orbitals cross (see figure 3(D)). Consequently the HOMO and LUMO orbitals hybridize strongly with each other. Their linear combination is symmetrical with respect to the central mirror plane (see the middle panel of figure 3(D)). Later, the HOMO and LUMO orbitals swap in the energy and an electron switching transfers the electron from LUMO to HOMO. Thus the anti-bonding orbital, the former LUMO state, is now occupied by two electrons while the bonding orbital is empty. This electronic configuration promotes the opening reaction.
100 fs, the system evolves such that the energy of HOMO/LUMO orbitals cross (see figure 3(D)). Consequently the HOMO and LUMO orbitals hybridize strongly with each other. Their linear combination is symmetrical with respect to the central mirror plane (see the middle panel of figure 3(D)). Later, the HOMO and LUMO orbitals swap in the energy and an electron switching transfers the electron from LUMO to HOMO. Thus the anti-bonding orbital, the former LUMO state, is now occupied by two electrons while the bonding orbital is empty. This electronic configuration promotes the opening reaction.
We note two interesting observations. First, in the case of the DAE–but molecule, we found two characteristic reaction times caused by different initial geometries. A rotation of the butane side groups around a single C−C bond in the side chain has a very low activation barrier. This gives rise to a large flexibility of the butane groups with two possible local minima configurations. In the first configuration, the butane groups are strongly rippled, almost perpendicularly with respect to the central switching unit, which significantly prolongs the reaction time of the opening process. In the second configuration, the butane groups are oriented almost straight along the central CHD unit, as shown in figure 2. Thus, the reaction lifetime is shorter but still longer than for a molecule gated to Au electrodes (see section 4) .
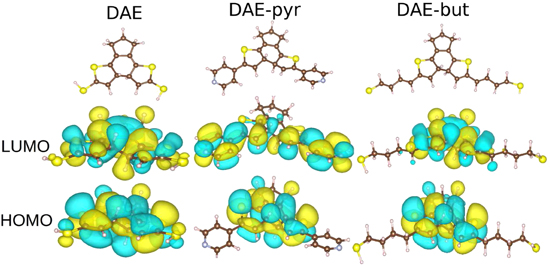
Figure 2. Atomic and electronic structure of DAE derivatives. The upper row represents three optimized atomic structures of the free-standing DAE derivates. The middle and low rows display the LUMO and HOMO states of the corresponding derivate.
Download figure:
Standard image High-resolution image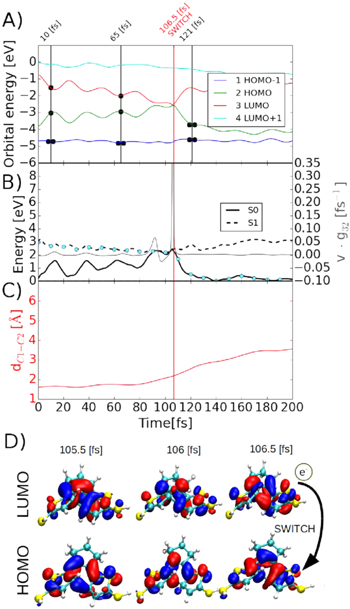
Figure 3. Analysis of the selected trajectory of the successful ring opening reaction of the DAE molecule. (A) Energy spectra for the four frontier molecular orbitals; the black dots indicate the electron occupancies of the different states. (B) Potential energy surfaces corresponding to the ground S0 and first excited S1 state together with the non-adiabatic coupling term  between HOMO and LUMO (solid grey line) [22]; the cyan dots indicate the actual PES for the simulation. (C) The distance between two interacting carbon atoms in the central ring of the DAE. (D) HOMO and LUMO orbitals in each of the three following MD steps right before the electron transfer.
between HOMO and LUMO (solid grey line) [22]; the cyan dots indicate the actual PES for the simulation. (C) The distance between two interacting carbon atoms in the central ring of the DAE. (D) HOMO and LUMO orbitals in each of the three following MD steps right before the electron transfer.
Download figure:
Standard image High-resolution imageSecond, surprisingly the DAE–but molecule has the lowest activation barrier in the excited state Eb = 0.03 eV between the considered molecules. Nevertheless the estimated reaction time is almost two times longer than for the other molecules. Again, we attribute this contradiction to the large flexibility of the butane side group. During the NEB [29] calculation of the activation barriers, two atoms on each end of the butane group were fixed, which almost impedes the rippling of the butane groups. However, this is not the case in NAMD simulations. This indicates that the rippling motion of the side groups significantly affects the opening reaction time. In other words, this indicates that not only the activation barrier but also attempted frequencies exciting a vibrational mode driving the C–C bond breaking determines the characteristic reaction times.
In summary, our NAMD simulations of the opening reaction for free-standing molecules gives a high quantum yield of the reaction. They also reveal no dependence on the localization of the LUMO orbital. The flexibility of the side groups significantly enlarges the reaction times even though the activation energy of the process is low.
4. Molecules bridging Au electrodes simulations
In the following, we discuss the non-adiabatic simulations of the extended system composed of the DAE derivatives bridging the gold electrodes.
In order to keep similar atomic configuration of the central DAE unit to the free-standing molecule, we optimized the electrode distance to reach a similar bond-length between two carbon atoms C1–C2 to the free-standing molecule. For this purpose, we carried out a set of relaxations of the molecules bridging electrodes for different electrode distances. This constraint introduces a slight compression strain in the junction. At the end of this section, we also analyze the effect of the mechanical strain on the switching mechanism for the DAE–Au system.
The electronic structure of the extended system with metallic electrodes consists of orbitals, which are localized either on the molecule, gold electrodes or both. The molecular HOMO and LUMO orbitals of the extended systems together with the projected density of state (PDOS) are plotted in figure 4. The HOMO orbitals are fully occupied. There is a slight difference between the localization of the HOMO of DAE, DAE–but and DAE–pyr, as shown in figure 4. The HOMO orbital of DAE–pyr is significantly below the Fermi level of the gold while the HOMO orbital of DAE and DAE–but are localized near the bottom edge near the Fermi level of gold. On the other hand, the LUMO orbitals are completely empty and located above the Fermi level of gold. The molecular band gap between the HOMO and LUMO orbitals in the DAE–Au and DAE–pyr–Au systems decrease with respect to the free-standing orbitals due to an additional screening of electrons due to the metallic electrodes [30]. In the case of DAE–but–Au, the screening effect is less pronounced due to a weak coupling of the HOMO/LUMO orbitals to the electrodes and the band gap remains almost unchanged compared to the free-standing molecule.

Figure 4. PDOS of all three DAE-derivatives between the gold electrodes for (A) DAE–Au, (B) DAE–pyr–Au, (C) DAE–but–Au. The blue lines express the PDOS on molecules and the black lines express the PDOS on gold electrodes. Molecular orbital HOMO and LUMO are plotted in real space above the graphs.
Download figure:
Standard image High-resolution imageThe electronic coupling (or spatial delocalization) of the LUMO molecular orbitals to the metallic electrodes is the most important feature of the system determining the feasibility of the opening reaction. The selected cases show different strengths of coupling. The LUMO molecular orbital of the DAE–Au system is mostly localized on the molecule with only a small contribution to the gold electrodes because of confinement between the frontiers gold atom and the molecule. In contrast, the LUMO orbital of the DAE–pyr–Au strongly hybridizes with the electronic states of gold. Thus it is delocalized over the whole system. Finally, the LUMO orbital of DAE–but–Au is only localized on the central part of the DAE unit, with negligible contribution to the but side groups and the Au electrodes, indicating only very weak electronic coupling between the molecule and electrodes.
To gain more insight into the reaction process, we also calculate the activation energy barriers of the reaction pathway of the extended systems in the ground state. In the case of DAE–Au the activation barrier is substantially lower with respect to the free-standing molecule. We can attribute this tendency to either the mechanical constraint induced by the gating or the strong electronic coupling of the HOMO orbital and the electrode. In contrast, the activation energy barrier for the DAE–but–Au and DAE–pyr–Au system is higher than for the free-standing molecules (see table 2).
Following on from this, we carried out an investigation on the electronic structure of gated molecules, and NAMD simulations of 100 trajectories with an initial geometry randomly chosen from the ground state molecular dynamic simulations previously calculated. The main goal of these simulations is to gain more insight into the electronic relaxation after the excitation during the reaction process. All simulations begin at a single excited state, where an electron is promoted from the HOMO to LUMO molecular orbitals shown in figure 4. In total, 25 (DAE–Au), 17 (DAE–pyr–Au) and 20 (DAE–but–Au) electronic states were propagated in time with the possibilities of electronic transitions between them. At the beginning, the LUMO molecular orbital is localized above several gold states, as can be seen from figure 5. Nevertheless the energy of the single occupied LUMO molecular orbital decreases rapidly from the beginning of the simulations crossing electronic states mostly localized on the electrodes. In the case of the DAE–Au and DAE–pyr–Au system we never observed in the simulations the complete opening reaction. This can be explained by the relatively strong coupling of the LUMO molecular orbital to electrodes, which causes almost immediate electron transfer from the LUMO orbital to the Au electrodes within  20 fs, as shown in figure 6. Once the electron is transferred into electronic states mostly localized on the electrode, the opening process is quenched. We should note that in our simulations, we never observed a transfer of the electron back to the LUMO orbital to complete the cyclo-reversion reaction. It indicates that the opening process is completely quenched once the electron is transferred from the LUMO molecular orbital to the electrode. This explains the low probability or even complete inhibition of the switching process observed experimentally [18].
20 fs, as shown in figure 6. Once the electron is transferred into electronic states mostly localized on the electrode, the opening process is quenched. We should note that in our simulations, we never observed a transfer of the electron back to the LUMO orbital to complete the cyclo-reversion reaction. It indicates that the opening process is completely quenched once the electron is transferred from the LUMO molecular orbital to the electrode. This explains the low probability or even complete inhibition of the switching process observed experimentally [18].
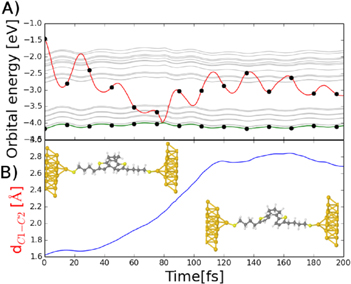
Figure 5. Analysis of the selected trajectory of successful ring opening reaction of DAE–but–Au. (A) Energy spectra for frontier molecular orbitals; the black dots indicate the electron occupancies of the different states. The green and red line corresponds to the LUMO and HOMO molecular orbital respectively. The gray lines are eigen energies of the gold states localized on gold electrodes. (B) The distance between two interacting carbon atoms in the central ring of the DAE.
Download figure:
Standard image High-resolution image
Figure 6. Analysis of the selected trajectory of unsuccessful ring opening reaction of DAE–but–Au. (A) Energy spectra for frontier molecular orbitals; the black dots indicate the electron occupancies of the different states. The green and red line corresponds to the LUMO and HOMO molecular orbital respectively. The gray lines are eigen energies of the gold states localized on gold electrodes. (B) The distance between two interacting carbon atoms in the central ring of the DAE–Au with an electrode distance of 11.44 Å.
Download figure:
Standard image High-resolution imageOn the other hand, the NAMD simulations of the DAE–but system revealed a different scenario. We detected 58 of 100 trajectories when the molecule was preserved in the excited state for a sufficient period, and the system underwent the opening reaction. The analysis of the successful cyclo-reversion reaction of the extended system with the DAE–but molecule is shown in figure 5. The LUMO molecular orbital of the DAE–but–Au is nicely localized on the switching unit and is well isolated from the gold electrodes by the anchoring groups. Again the simulation starts by the rapid decreasing of the orbital LUMO energy crossing orbitals localized on the electrodes without any electron transition to electrodes. The energy of the LUMO orbital approaches the energy of the HOMO orbital. They swap in energy at  80 fs and the anti-bonding, former LUMO, orbital remained still occupied by one electron. In this situation, the close configuration becomes unstable and the C1–C2 bond breaks.
80 fs and the anti-bonding, former LUMO, orbital remained still occupied by one electron. In this situation, the close configuration becomes unstable and the C1–C2 bond breaks.
The result of the NAMD simulations shows the strong correlation between the opening mechanism and the delocalization of the LUMO molecular orbital, where an electron is excited. We calculated the spatial distribution [31] of the LUMO orbital to quantitatively analyze its delocalization for the considered systems for each initial geometry of the NAMD simulations. The number of accessible atoms by the LUMO orbitals with respect to the C1–C2 bond length for all the initial configurations is plotted in figure 7.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 7. The localization of the LUMO orbitals of the initial structures used in simulations in dependence on the initial bond length C1–C2. Fill marks correspond to the molecules which remain closed. Empty marks correspond to the molecules which transfer to the open position. The top graph shows the results for molecules under any tension. The distances of the electrodes were of DAE–Au, DAE–pyr–Au, DAE–but–Au were: 11.44 Å, 16.46 Å, 23.15 Å. The lower graph shows the results of the DAE molecule under tension between electrodes which are 14.04 Å
Download figure:
Standard image High-resolution image{kind=link}
Finally, we analyze the impact of the mechanical stress on the switching rates in the case of the DAE–Au system. Particularly, we increased the distance between the electrodes by 11% and 23% respectively with respect to the previous case. Consequently the bond-length C1–C2 elongates as well (see table 3). For the elongation of 11% the molecular DAE–Au junction is found in the equilibrium position with the minimum total energy, free of the mechanical stress. Further elongation of the electrode distance introduces the tensile strain. Surprisingly, the activation energy barrier in the ground state changes non-monotonically with the bond-length C1–C2, reaching the maximum value of 0.9 eV for the junction in the equilibrium distance without any mechanical stress. On the other hand, the NAMD simulations reveal completely different behavior of the switching mechanism when the system is driven into the excited states. The success ratio of the opening process drastically changes from 0 to almost 50% when the electrode distance is elongated from the compressed to the equilibrium configuration of the whole molecular junction. On the other hand, the C1–C2 distance changes only by 0.01 Å. Thus we attributed this effect to the electronic coupling of the LUMO orbital to the electrodes, which facilitates the electron transfer from the LUMO orbital to the electrodes and consequently quenches the reaction process.
Table 3. Distances between gold electrodes and interacting carbon atoms. Quantum yield of the reaction, barrier height Eb of the reaction calculated from the nudge elastic band simulations and reaction times of the ring opening reactions.
 [Å] [Å] |
C1–C2 [Å] | open/close | Eb [eV] | treac [fs] |
|---|---|---|---|---|
| 11.44 | 1.59 | 0/100 | 0.5 | — |
| 12.74 | 1.60 | 47/53 | 0.9 | 295.3 |
| 14.04 | 1.65 | 57/43 | 0.7 | 355.28 |
The quantum yield of the reaction is further increased by about 10% when the tensile stress is applied. The quantum yield under tension is about 57% but still is much lower than for the free-standing DAE molecule, being 90%. We can conclude that the external mechanical stress has significant impact on the reaction process. In particular, when the junction is submitted to the compression stress, it enhances the electronic coupling of the LUMO orbital to the electronic states of the electrodes. In principle, this phenomenon can be used for the design of the mechanical sensor on a single molecule scale.
5. Conclusion
In this paper we have studied the cycloreversion reaction of free-standing and gated DAE derivates with different anchoring groups by NAMD simulations. For the first time, we show that NAMD simulations of complex systems with metallic electrodes, including hundreds of atoms, is feasible. We analyzed the quenching of the photochoromic opening reaction of the molecules placed between metallic electrodes. We identify that the quenching mechanism is driven by the strong electronic coupling of the LUMO molecular orbitals to electronic states of the metallic orbitals. We also demonstrate that the electronic coupling can be tuned by externally applied mechanical stress, which can be adopted for the development of mechanical sensors on a single-molecule scale.
Acknowledgments
This work was financially supported by a Czech Science Foundation grant no. 14-02079S.