Abstract
The 229thorium isotope presents an extremely low-energy isomer state of the nucleus which is expected around 7.8 eV, in the vacuum ultraviolet (VUV) regime. This unique system may bridge between atomic and nuclear physics, enabling coherent manipulation and precision spectroscopy of nuclear quantum states using laser light. It has been proposed to implant 229thorium into VUV transparent crystal matrices to facilitate laser spectroscopy and possibly realize a solid-state nuclear clock. In this work, we validate the feasibility of this approach by computer modelling of thorium doping into calcium fluoride single crystals. Using atomistic modelling and full electronic structure calculations, we find a persistent large band gap and no additional electronic levels emerging in the middle of the gap due to the presence of the dopant, which should allow direct optical interrogation of the nuclear transition.
Based on the electronic structure, we estimate the thorium nuclear quantum levels within the solid-state environment. Precision laser spectroscopy of these levels will allow the study of a broad range of crystal field effects, transferring Mössbauer spectroscopy into the optical regime.
Export citation and abstract BibTeX RIS
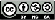
Content from this work may be used under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
1.1. The amazing 229Th isomer state
The radioisotope 229thorium (lifetime 7932 years, [1]) is expected to have a long-lived, exceptionally low-energy, isomer state—probably the lowest of all nuclear levels. The currently most accepted value for its energy is 7.8 eV, obtained in an indirect gamma-spectroscopy measurement [2, 3], placing it into the vacuum ultraviolet (VUV) range (approximately 160 nm). The possibility of manipulating the quantum states of a nucleus with laser light has engaged researchers from various fields for more than 30 years. Possible applications range from a nuclear frequency standard [4–7] to gamma-ray lasers [8].
All of these exciting applications obviously require exact knowledge of the energy and lifetime of the isomer state, which currently remains elusive. The predicted isomer energy has undergone a series of changes over the years: from 3.5 eV [9] to 5.5 eV [10], and to 7.8 eV [2, 3], in the VUV range. Conclusions drawn from the experimental results and even the mere existence of the isomer state are still under discussion [11].
Predictions for the isomer lifetime are equally scattered: [12] yields a theoretical prediction of T1/2 = (10.95 h)/ (0.025E3), where E is the isomer transition energy in eV, which leads to 55 min for E = 7.8 eV . Several experiments have been performed, proposing lifetimes between 2 min [13] and 6 h [14]. These results are heavily disputed [15].
A spectroscopy experiment to determine these values should therefore be able to cover a large range of wavelengths and be compatible with a maximum number of excitation sources and measurement schemes. Furthermore, the exact chemical composition of the thorium ions needs to be known and controlled, as it may have a crucial effect on the isomer lifetime and the nature of the de-excitation process [16].
1.2. The solid-state approach
It was first suggested in 2003 by Peik et al [4] that thorium ions may be implanted into UV transparent crystals to perform laser spectroscopy of the isomer state and eventually realize a nuclear clock. Due to shielding by the electron shell, the favourable properties of the nuclear transition (narrow line width) could be maintained, opening the possibility of building a solid-state optical clock [6, 7]. Due to the huge mass of the nucleus (compared to electrons), the nuclear transition would lie deep in the Lamb–Dicke regime, decoupling internal and external degrees of freedom to first order.
Although inhomogeneous effects [7] may ultimately hinder the design of high-performance solid-state optical clocks, such thorium-doped crystals can be a promising system for an initial 'coarse-grained' measurement of the isomer transition energy. Moreover, crystal effects (including temperature and pressure dependences) can be probed directly with a laser, transferring Mössbauer spectroscopy into the optical regime. Such investigations not only allow studying the host crystal properties but could give additional information about magnetic dipole and electric quadrupole momenta of the thorium isomer nucleus.
1.3. Calcium fluoride
The host material for thorium doping obviously has to be transparent in the wavelength region in which the nuclear transition is to be investigated. Furthermore, it has to chemically accept the dopant in sufficient concentration. The Th-doped material must remain transparent, which means a sufficiently large band gap and no additional electronic levels emerging, which might quench the nuclear transition or inhibit excitation.
In our investigation, we have chosen calcium fluoride (CaF2) single crystals as a host material for thorium doping. Other possible host systems, in particular LiCaAlF6, have been considered by the group of Hudson [17]; an overview of crystal candidates can be found in [18]. CaF2 is a standard material in UV optics due to its wide band gap. The literature values range from 11.6 eV [19] to 12.1 eV [20, 21] for a direct gap transition. The valence band of CaF2 consists of 2p levels of the fluorine ions, while the bottom of the conduction band originates from the 4s and 3d orbitals of the calcium ions. CaF2 presents a high radiation damage threshold, it can be grown to high quality using a multitude of established techniques such as Czochralski, Bridgman and micro-pulling; and cutting, polishing and coating techniques are readily available, even for large volume crystals.
The simplelattice structure of CaF2 calls for ab initio calculations as performed in this work. Doping of CaF2 has been studied extensively (both theoretically and experimentally) in the context of laser materials, focusing on trivalent dopants, exhibiting strong optical activity caused by electronic transitions. Due to the high electropositivity of thorium (1.3 Pauling), we expect to find the dopants in the Th4+ state, leading to a radon-like noble gas electron configuration. The ionic radii of Ca2+ and Th4+ are 1.26 Å and 1.19 Å, respectively [22]; their similarity should facilitate replacing a Ca2+ by Th4+ in the doping process.

Figure 1. Undoped CaF2, showing the simple cubic unit cell containing four formula units. Ca ions are blue, and F ions are yellow.
Download figure:
Standard image High-resolution image2. Computer modelling
In this paper, both atomistic modelling and density functional theory have been used, as they provide complementary information.
The combination of the empirical GULP method and ab initio calculation has the double advantage of providing us with defect formation energies and solution energies as well as precise determination of the electronic structure, giving a reasonable size of the band gap and yielding reliable energies for the impurity states. Although it is in principle possible to derive defect formation energies and solution energies using ab initio calculations, the GULP method provides this information with a massively reduced computational effort and with the required accuracy.
2.1. Atomistic modelling: GULP
The GULP code (General Utility Lattice Program) is a general-purpose atomistic modelling code [23]. It uses effective interatomic potentials to calculate the lattice and defect properties of materials. The potential model used is the Buckingham form, supplemented by an electrostatic term:
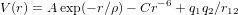
where A, ρ and C are parameters whose values are specified for each interaction between a pair of ions, and q1 and q2 are the charges on the interacting ions. The potential parameters are given in table 2.
In order to carry out defect calculations, the Mott–Littleton approximation is used, in which the region immediately surrounding the defect is modelled explicitly, and approximations are used for more distant regions of the lattice [24]. Region I is the region immediately surrounding the defect, and a consistent value of 10 Å was used in these calculations for its radius. Region IIA is the interface region, with a radius of 20 Å.
2.2. Ab initio modelling: VASP
CaF2 can be represented as a simple cubic lattice with space group Fm3m (225) (see figure 1). The Ca atoms occupy the 4a positions and the F atoms the 8c positions. Ca is surrounded by eight F atoms, whereas F sits in the centre of a Ca tetrahedron.
To calculate the electronic structure of Th-doped CaF2 we assume a supercell based on the rhombohedral primitive cell. The impurity problem is simulated by assuming a periodically repeated cell containing 27 formula units (81 atoms). By replacing one Ca by Th we simulate an effective impurity concentration of 3.7% per Ca.
To calculate the electronic structure, we employ the Vienna ab initio simulation package (VASP 5.2/5.3) method, as devised by Kresse and Furthmüller [25]. Since for semiconductors and insulators there are well-known deficiencies of the local spin density approximation (LSDA) or the spin-polarized generalized gradient approximation (SGGA) in determining the proper gap size [26–28] and the position of the impurity states [29], we perform our calculations using the hybrid Hartree–Fock density functionals in the HSE06 version [30, 31]. To reduce the computation time, we relax the cell size and the ionic positions within a PBE [32] calculation. The geometries were relaxed until all force components were less than 0.01 eV Å−1. The relaxed lattice constant is only 0.88% larger than the experimental value, which gives confidence in the accuracy of the method. After convergence we keep these relaxed coordinates and change to the HSE functional. All results are obtained using the projector augmented plane wave method [33, 34] by explicitly treating 12 valence electrons for Th (6s2, 6p6, 7s2, 6d2), eight valence electrons for Ca (3p6, 4s2), seven valence electrons for F (2s2, 2p5) and six valence electrons for O (2s2, 2p4), respectively. In order to avoid Pulay stress and related problems, plane waves up to a cut-off energy of 500 eV were included in the basis set. The Brillouin zone integration was carried out over a 2 × 2 × 2 Γ-centred Monkhorst–Pack k-mesh with a Gaussian broadening of 0.02 eV. The total energy was converged to 1 × 10−6 eV .
3. Modelling results
3.1. GULP results
These calculations were performed using the GULP code, as discussed in section 2.1.
3.1.1. Potentials and structural agreement
The CaF2 structure was taken from [35], and table 1 shows the agreement between experimental and modelled structures. The potential parameters employed in the study are given in table 2.
From table 1 it is seen that the lattice parameters are reproduced to better than 1.5%, giving confidence in the use of the potential for the defect calculations reported in section 3.2.
Table 1. Experimental and calculated lattice parameters.
| Lattice parameter | Experimental (Å) [34] | Calculated (Å) | % difference |
|---|---|---|---|
| a | 5.47 | 5.55 | 1.47 |
| b | 5.47 | 5.55 | 1.47 |
| c | 5.47 | 5.55 | 1.47 |
Table 2. Potential parameters employed in the study. (Note: F shell parameters: shell charge (Y ) = −1.378 |e|, spring constant (k) = 24.36 eV Å−2.)
| Interaction | A (eV ) | ρ (Å) | C (eV Å6) |
|---|---|---|---|
| Ca–F | 3400.0 | 0.2664 | 0.0 |
| F–F | 911.69 | 0.2707 | 13.80 |
| Th–F | 3500.0 | 0.2980 | 0.0 |
3.1.2. Intrinsic defects in CaF2
Calculations of basic intrinsic defects (vacancies and interstitials) have been performed. In table 3 the defect formation energies are given, and in table 4 they are combined to give Frenkel and Schottky formation energies. The lattice energy of CaF2, needed in the Schottky defect formation energy calculation, is −26.95 eV.
Table 3. Basic defect formation energies.
| Defect (fractional coordinates) | Formation energy (eV) |
|---|---|
| Ca vacancy (0.0, 0.0, 0.0) | 24.20 |
| Ca interstitial (0.5, 0.0, 0.0) | −13.48 |
| F vacancy (0.25, 0.25, 0.25) | 5.08 |
| F interstitial (0.5, 0.0, 0.0) | −1.90 |
Table 4. Frenkel and Schottky energies.
| Defect (fractional coordinates) | Formation energy per defect (eV) |
|---|---|
| CaF2 Schottky (unbound) | 2.53 (experimental 2.70 in [36]) |
| Ca Frenkel | 5.46 |
| F Frenkel | 1.59 |
From table 4 it is clear that the preferred form of intrinsic disorder is F Frenkel, suggesting that  interstitials may be present, which is an important conclusion for later in the paper when thorium doping is considered.
interstitials may be present, which is an important conclusion for later in the paper when thorium doping is considered.
3.1.3. Thorium doping calculations
When the CaF2 structure is doped with Th4+, it is reasonable to assume that the Th4+ ion will substitute at the Ca2+ site in the structure (see figure 2). The calculated substitution energy is −43.44 eV. When the doping takes place, there is a charge imbalance in the lattice of +2, so charge compensation has to be involved. Two mechanisms were considered.

Figure 2. Charge compensation after Th doping (Th4+ ion in red). (a) compensation by a Ca2+ vacancy, (b)–(d) compensation by interstitials (green).
Download figure:
Standard image High-resolution image3.1.4. Charge compensation by Ca2+ vacancy formation
The equation for the doping process may be written as
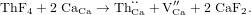
Here, standard Kröger–Vink notation has been used for the defects [37].
The solution energy is then obtained from
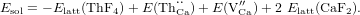
Unbound and bound values have been obtained, with the bound values calculating the thorium doping and calcium vacancy formation together as a defect cluster (giving a formation energy of −22.74 eV). These are given in table 5.
3.1.5. Charge compensation by  interstitial formation
interstitial formation
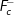
The equation for the doping process may be written as
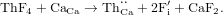
The solution energy is then obtained from
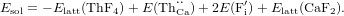
Since two interstitials are formed, their respective location needs to be considered when bound solution energies are calculated. Three possible configurations have been analysed: (i) linear, (ii) 90° and (iii) 125.3°, as illustrated in figures 2(b)–(d).
Table 5. Solution energies for thorium doping. (Note: ThF4 lattice energy =−79.98 eV.)
| Configuration | Solution energy per defect (eV) |
|---|---|
| Vacancy compensation, unbound | 3.42 |
| Vacancy compensation, bound | 1.67 |
| Interstitial compensation, unbound | 2.90 |
| Bound interstitial—180° | 0.47 |
| Bound interstitial—90° | 0.32 |
| Bound interstitial—125.3° | 0.67 |
The clear conclusion is that interstitial compensation is most favourable in this case, and further, that the configuration where the two F interstitials are at 90° is of lowest energy. Note that the solution energies calculated with the GULP code allow us to compare different stoichiometries.
3.2. VASP results
CaF2 is a wide gap insulator; its electronic structure is well described within the applied ab initio method. For pure CaF2 we calculate an effective gap of about 9 eV (see figure 3 and table 6), which is in fair comparison with the experimental value of 12 eV. It is also known that for these wide gap insulators advanced post-DFT functionals such as HSE06 tend to underestimate the gap size. However, the positions of the impurity bands are scaled down accordingly, which makes it straightforward to compare our calculated results with experimental values. The valence band consists of completely filled 2p states of the F− ions with a small admixture of Ca2+ states. The conduction band contains the empty s and d states of Ca2+, again with a small admixture of F states. The density of states (DOS) of undoped CaF2 is shown in figure 3.

Figure 3. Density of states of undoped CaF2 per supercell containing 27 formula units.
Download figure:
Standard image High-resolution image3.2.1. Charge compensation by interstitial formation
Substituting one Ca by Th means that the tetravalent Th4+ provides two additional electrons which would occupy states in the conduction band, leading to a metallic state. To remain an insulator, these two electrons need to be bound to additional atoms that provide charge compensation. Our GULP study showed that an F Frenkel defect is energetically most favourable, which means that the presence of two additional ions will provide the necessary charge compensation; see table 5. Here, we simulate three different configurations for these two ions, forming 180°, 90° and 125.3° angles with respect to the Th impurity. While a simple electrostatic argument would point to the 180° configuration being most favourable, our calculation finds the 90° geometry as a ground state, which confirms the GULP results.
The insertion of Th4+ and the two additional ions leads to relaxations of the crystalline lattice, which are shown in figure 4. The main effect is a movement of Th4+ and the two ions towards each other. The presence of the negatively charged charge compensation ions exerts a repulsive force on the neighbouring F lattice ions, pushing them outwards.

Figure 4. Schematic view of the forces acting on the atoms in Th-doped CaF2 in the 90° F charge-compensated case: Th (red), F (yellow), charge compensation (green), Ca (blue). The arrows point in the direction of deformation and the length of the arrow is proportional to the force. The relaxations are of the order of 5–10% of the bond length.
Download figure:
Standard image High-resolution image
Figure 5. Charge density for the 90° F-interstitial charge compensation. Left panel: in the (110)-plane, comprising the Th dopant and the two interstitials that provide the charge compensation. Right panel: charge density cut in the  plane.
plane.
Download figure:
Standard image High-resolution imageThe charge density in figure 5 shows in the left panel a section through the plane spanned by the Th and the two ions (110) in the 90° configuration. The ions move slightly towards the Th, which is also reflected by the increase in charge density between Th and . The right panel is a section in the plane, and it clearly shows the outward movement of the next-neighbour lattice F ions.

Figure 6. Density of states for Th-doped CaF2 with two ions for charge compensation in the 90° configuration (see table 6, case 2 and figure 2(b)). The densities of states of the two additional ions are at the bottom of the gap; the empty Th 5f states are located at about 8.59 eV, slightly reducing the gap.
Download figure:
Standard image High-resolution imageFigure 6 shows the density of states for the Th-doped and 90° charge-compensated case. The electronic states of the two additional ions are located at the bottom of the gap, forming a narrow impurity peak. Th, which replaces a Ca site, is in a 4+ state, so the empty Th states form the conduction band together with the unoccupied Ca states. The unoccupied Th 5f states form a narrow band at the top of the insulating gap, reducing it to about 9 eV. Although we observe a reduction of the band gap upon Th doping (5.5% compared to the undoped case), the band gap remains large enough to be transparent for the 7.8 eV optical excitation. Both other configurations (180°, 125.3°) exhibit an almost identical density of states; the small energetic differences do not appear.
3.2.2. Charge compensation by Ca2+ vacancy formation
A different route of charge compensation is the creation of a Ca2+ vacancy, as discussed in 3.1.4. The respective density of states looks almost identical to the compensated case, but without the additional states at the bottom of the gap. Our GULP analysis (see table 5) gives a defect formation energy of 1.67 eV, which is more than five times larger than the energy involved in the creation of two interstitial ions. We therefore consider this configuration as unlikely.

Figure 7. Density of states of Th-doped CaF2 with a Ca2+ vacancy for charge compensation. The missing Ca atom reduces the DOS of the empty states above the gap; the Th 5f states are at 8.33 eV (compare figure 6).
Download figure:
Standard image High-resolution image3.2.3. Oxygen impurities
Since in the crystal production process oxygen could be easily present as an impurity, we also studied compensation mechanisms with oxygen involved. These is one  at an interstitial position, there are two O− on two F positions and there is a mixture of an O− on an F position and an interstitial (see figure 8). In all cases we find oxygen states appearing inside the gap in the density of states, as is shown in figure 9. The presence of O reduces the gap to about 6 eV (see table 6, cases 6–8), making it unfavourable for the desired optical excitation. Hence, oxygen impurities have to be avoided in the crystal fabrication process.
at an interstitial position, there are two O− on two F positions and there is a mixture of an O− on an F position and an interstitial (see figure 8). In all cases we find oxygen states appearing inside the gap in the density of states, as is shown in figure 9. The presence of O reduces the gap to about 6 eV (see table 6, cases 6–8), making it unfavourable for the desired optical excitation. Hence, oxygen impurities have to be avoided in the crystal fabrication process.

Figure 8. Charge compensation after Th doping (Th4+ ion in red). (a) Compensation by an O2− impurity interstitial (violet), (b) compensation by two O− impurity ions (violet), substituting F ions, (c) compensation by one O− ion substituting an F ion (violet) and an interstitial (green).
Download figure:
Standard image High-resolution image
Figure 9. Density of states for Th-doped CaF2 with O2− at an interstitial site (see table 6, case 6). The additional O2− ion forms a state within the gap, thus reducing it to 6.25 eV.
Download figure:
Standard image High-resolution imageTable 6 shows the main results for the eight different cases studied. Since within VASP a comparison of the total energies makes sense only for the three stoichiometrically identical charge compensations (rows 2–4), for all other cases the total energies are omitted. The coordinates are given for the unrelaxed positions with respect to Th in multiples of the lattice constant. It should be noted that due to the relaxation of the atomic positions the true coordinates and angles differ slightly.
In the first row we give the band gap for the undoped CaF2. The next three rows are the different configurations for the two additional fluorine atoms at interstitial positions. The 90° configuration yields the lowest total energy, which is set to zero. The two other configurations are higher in energy. The next three rows give the results for a Ca vacancy (row 5) and two types of O impurities at different positions, O at an interstitial site (row 6) and two O replacing two F atoms (row 7). Row 8 gives the result for a mixed configuration with one O at an F site and an additional interstitial.
Table 6. Calculated band gaps, total energies and electric field gradients for the different configurations investigated in the fully relaxed structure.
| Case # | Charge compensation | Angle (deg) | Relaxed energy (eV cell) | Band gap (eV) | |
|---|---|---|---|---|---|
| 1 | Undoped CaF2 | 9.07 | |||
| 2 |  |
 |
90 | 0 | 8.59 |
| 3 | |
 |
180 | 0.42 | 8.07 |
| 4 |  |
 |
125.3 | 0.52 | 8.00 |
| 5 |  |
8.33 | |||
| 6 |  |
6.25 | |||
| 7 |  |
 |
109.5 | 6.68 | |
| 8 | |
|
54.7 | 7.07 | |
4. Solid-state–nuclear interactions
The interaction between the crystal lattice and the thorium nucleus leads to the appearance of hyperfine structure in the nuclear quantum levels. In an ionic crystal, fine structure is absent due to the lack of unpaired electrons. The Hamiltonian of such hyperfine interaction can be represented in the form HHFS = HE0 + HM1 + HE2 + ⋯ [6]. A general discussion of these terms and various inhomogeneous effects is given in [7]. Here, we focus our attention on the homogeneous effects, i.e., details of the hyperfine structure that are common and specific for certain configurations of charge compensation.
The electric monopole shift or isomer shift (HE0) arises from contact interaction between the electron cloud and the finite volume of the nucleus [38]:
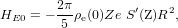
where ρe(0) is electron charge density near the nucleus calculated in non-relativistic approach, Ze and R are the charge and radius of the nucleus, respectively (here, the nucleus is supposed to be spherical; otherwise, we should replace R2 by 5r2/3, where r is the RMS radius of the nuclear charge distribution), and S'(Z) is the relativistic correction (for thorium, S'(90) = 11.7 [38]). The nuclear radii for both the ground state and the isomer state of 229Th have been theoretically calculated in [39].
In our ab initio calculation, the core electrons are supposed to have a fixed, environment-independent electron density. However, contributions from the valence electrons may change for the different charge compensation configurations discussed above. This effect leads to a shift of isomer transition energy,
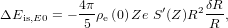
where δR = Ris − Rg is the difference between the characteristic radii of the thorium nucleus in the isomeric state and the ground state, respectively. Unfortunately, the quantity ρe(0) is not yet accessible in VASP. Even if the electron density of the valence electrons at the position of the Th nucleus could be extracted with confidence, it is questionable whether interactions with the deeply bound core electrons (not explicitly modelled in VASP) could be neglected.
For an order of magnitude estimate of the isomer shift we therefore look at results from Mössbauer spectroscopy of 151Eu2+ doped into CaF2. Here, dependence of the isomer shift on the doping concentration was observed [40], and it was conjectured to be an effect of a change in lattice constant [41]. Scaling these results to the thorium-doped system and using the RMS charge radius from [39, 42], we obtain an estimate of a few GHz for the isomer shift between the different charge compensation configurations. This is to be considered a very coarse estimate, as especially the theoretical value for the nuclear charge radius is strongly model-dependent.
The mean value of the magnetic dipole interaction term (HM1) goes to zero. At the same time, interaction of the thorium nucleus with fluctuating magnetic fields of surrounding fluorine nuclei leads to an inhomogeneous broadening of the isomer transition; see [7].
The Fermi contact interaction between the valence electrons and the thorium nucleus vanishes because the valence electrons are also spin paired.
The electric quadrupole interaction term HE2 can be written as [43]
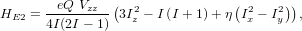
where eQ is the nuclear quadrupole momentum, Ix, Iy and Iz are the components of the nuclear angular momentum, I is its value, Vzz is the maximum main value of the electric field gradient and η = Vxx − Vyy /|Vzz| is the asymmetry. Within the VASP ab initio calculation the elements of the electric field gradients are calculated directly from the non-spherical part of the potential, following the method devised by Petrilli et al [44].
The quadrupole interaction leads to a splitting of the ground and the isomeric energy levels, as shown in figure 10.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 10. Quadrupole structure of the ground and isomer states of the 229Th nucleus in the presence of a non-zero magnetic field gradient and notation for energy corrections (see table 7). Black rectangles denote the eigenstates of Iz giving the main contribution to the corresponding energy state. Grey rectangles illustrate the fact that admixtures of other magnetic states contribute to the energy state for η≠0.
Download figure:
Standard image High-resolution image{kind=link}
The most recent experimental value for the quadrupole moment of the ground state is Qg = 3.11(16) b [45]; the quadrupole moment Qis of the isomer state is still unknown: estimations yield Qis ≈ 1.8 b [8]. Values of the quadrupole shifts for different sublevels calculated using the VASP data are tabulated in the table 7.
We believe that different doping configurations (if present in a crystal) could be identified in spectroscopy by their isomer shift and quadrupole structure. Studies of this quadrupole structure will allow determination of the nuclear quadrupole moment of the isomer state. Note that, as the transition probabilities can be calculated easily, the relative intensities are known [7], facilitating identification. Transitions within the J = 5/2 ground state are also accessible to NMR spectroscopy.
Table 7.
Calculated electric field gradient and asymmetry factor η and quadrupole shifts of ground and isomer levels of the 229Th nucleus corresponding to different charge compensation configurations.  .
.
| Charge compensation | Elect. gradient | Shifts of quadrupole sublevels (MHz) | ||||||
|---|---|---|---|---|---|---|---|---|
| First ion | Second ion | Angle (deg) | Vzz (V Å−2) | η | E1/2 | E3/2 | E5/2 |  |
| |
|
90 | 223.0 | 0.48 | −370 | −58 | 420 | −250 |
| |
|
180 | −296.7 | 0 | 450 | 110 | −560 | 320 |
| |
 |
125.3 | −265.5 | 0.2 | 410 | 94 | −500 | 290 |
| |
— | — | 68.3 | 0 | −100 | −26 | 130 | −74 |
| |
— | — | −10.4 | 0 | 16 | 3.9 | −20 | 11 |
| |
 |
109.5 | −18.4 | 0.02 | 28 | 6.9 | −35 | 20 |
| |
|
54.7 | 31.8 | 0.37 | −51 | −9.7 | 60 | −35 |
5. Conclusions
We have theoretically analysed thorium-doped calcium fluoride using two complementary methods: atomistic modelling based on the GULP code and ab initio simulations based on VASP. Thorium is expected to replace Ca2+ ions in the crystal lattice as Th4+. We show that the most probable mechanism of charge compensation is the introduction of two interstitials in a 90° configuration.
We calculate the local lattice deformation around the dopant and the resulting electric field gradient. Interactions of the nuclear quadrupole with this gradient leads to a splitting of the isomer levels, and measuring the quadrupole shifts in optical or NMR spectroscopy will allow us to corroborate our assumptions on the microscopic doping configuration. We show that the doping leads to a reduction of the material band gap by only a few per cent. A search for the 229thorium nuclear transition energy is hence possible from the optical region until deep into the UV region, validating the approach of nuclear laser spectroscopy in a solid-state sample.
Acknowledgments
GK was supported by the FWF project M1272-N16 'TheoNAC' and the WPI Thematic Program 'Tailored Quantum Materials'. PD, GW, MS, GK, and TS acknowledge support by the FWF Project Y481 and the ERC project 258604-NAC. PD and PM acknowledge support from the Austrian Science Fonds FWF within the SFB ViCoM F4109-N13 P09. GW and MS further thank the FWF doctoral program on 'Complex Quantum Systems'. All authors acknowledge discussions with E Hudson, W Rellergert, and R Uecker on crystal doping and with E Litvinova on the thorium nuclear charge radius.