
Abstract
The optical constants together with the magneto-optical Voigt constants of several phthalocyanine (Pc) and methoxy functionalized tetraphenylporphyrin (TMPP) thin films prepared on silicon substrates are presented. The materials investigated are MePc with Me = Fe, Co, Ni, Cu, Zn and MeTMPP with Me = Cu, Ni. We also compared our results to the metal-free H2Pc, H2TPP and H2TMPP. The experimental results will be supported by electronic structure calculations based on density functional theory (DFT) and interpreted using the perimeter model initially proposed by Platt. The model allows for qualitative understanding of the forbidden character of transitions in planar, aromatic molecules, and is able to qualify differences between Pc and TMPP type materials.
Export citation and abstract BibTeX RIS
1. Introduction
Since the first reports of an organic spin valve [1, 2], novel devices such as spin-OLEDs (organic light emitting diodes) [3] and spin-OFETs (organic field effect transistors) [4] as well as sensors based on electrically detected magnetic resonance [5] have been developed. The rapid development of the field of organic spintronics is driven by the large spin life time in organic molecules, combined with the great diversity and flexibility of molecular synthesis and technological processing. The large range of molecules which has been integrated into spintronic devices to date comprises various diamagnetic molecules, e.g. [6], and individual single molecule magnets [7].
Among others, the groups of phthalocyanines (Pcs) and methoxy functionalized tetraphenylporphyrins (TMPPs) are very promising in terms of application in organic electronics and spintronics, due to their chemical and thermal stability and the possibility of tuning their electronic and magnetic properties by changing the central metal atom or substituting H-atoms of the ligands with functional groups. We present also TMPP results, since they have not been characterized with regard to different metal centers until now. Very recently an additional way to further engineer the magnetic properties by building phthalocyanine heterojunctions was discovered [8]. The possibility of producing well ordered films by thermal evaporation in vacuum makes them perfect model systems for preparing device-like structures.
To understand the charge and spin transport properties of a material it is essential to know the electronic structure around the Fermi energy since typically only the few uppermost occupied and lowest unoccupied electronic levels define the transport characteristics. Therefore a clear understanding of the optical properties is an essential ingredient to reveal the electronic structure of a material. Additionally the anisotropy of the optical constants along different space directions can be used to obtain information about structural parameters of the sample [9, 10].
Reports on magneto-optical studies conducted by magnetic circular dichroism (MCD) spectroscopy in the typical energy range of electronic transitions of the porphyrinoids3 are numerous. Reference [11] provides an extensive overview on MCD measurements and spectral deconvolution analysis for several porphyrinoids. Early MCD spectral deconvolutions for porphyrinoids were performed in [12, 13]. From the lineshape of the MCD bands conclusions were drawn regarding the nature of the ground and excited states of the most prominent absorption bands of H2Pc, CoPc, NiPc and CuPc. However, the commonly used absorption spectroscopy and MCD require the material to be dissolved in a liquid solvent or to be prepared as a film on transparent substrates. In this work films on silicon substrates have been investigated, which is much closer to the condition used in organic electronics and spintronics devices.
The applied variable angle spectroscopic ellipsometry allows us to detect the anisotropy between the optical constants for light polarization in the plane of the film and the polarization perpendicular to the film plane. The magneto-optical Kerr effect (MOKE) spectroscopy setup used measures the full complex Kerr angle, i.e. both Kerr rotation and Kerr ellipticity were obtained. However, the shape of the raw MOKE spectrum is thickness dependent [10] due to optical interference within the film. Therefore, in contrast to MCD results, the MOKE data require numerical post-processing before they can be interpreted. From the measured complex MOKE spectra and from the optical constants determined from ellipsometry, the material-specific magneto-optical Voigt constant can be obtained [10]. The Voigt constant Q = −iεy/εx is part of the off-diagonal elements of the dielectric tensor in a magnetic field along z. The Voigt constant is material specific and the magnitude and lineshape of the features occurring in its energy dispersion spectrum in the near infrared to near ultraviolet spectral range can be used to extract information on the spin and/or orbital degeneracy as well as on the symmetry of the electronic levels involved in the measured electronic transitions. Moreover, with the knowledge of the complex Voigt constant, MCD as well as Faraday spectra can be predicted.
The perimeter model is a qualitative model, which can be used for the characterization of the forbidden character of π→π∗ transitions in planar, aromatic molecules. More specifically, it requires the π-system of interest to contain (4N + 2) electrons, which is the Hückel condition for aromaticity. The main point of the model is to approximate planar, aromatic molecules of arbitrary shapes by a 1D-quantum mechanical oscillator along the circumference of an appropriately chosen ring. It was first demonstrated by Platt in 1949 [14] and used for simple aromatic molecules such as benzene or anthracene. The idea behind it might be clarified by taking benzene as a small example. Benzene with its six delocalized π-electrons is a perfectly planar and regular hexagon which is replaced in the perimeter model by six free electrons in a 2D-disk. From this one can finally define hypothetical orbital angular ring quantum numbers (OARNs), which can be used in selection rules for possible excitations in the system.
As shown in [15–19] this model makes valid predictions on the character of weakly dipole forbidden transitions. Even for porphyrins [20, 21] and derivates [22] the model could be applied with partial success. The prediction of OARNs requires only the knowledge of the real-valued representations of the molecular wavefunctions, which are existing whenever no external electric or magnetic field is present. Thus we would like to show how this electronic structure calculation information can be transformed by means of this simple model into useful data.
In section 2 we present details on the experimental methods and section 3 outlines the theoretical background. The last two sections 4 and 5 contain the discussion and the summary of our results.
2. Experimental details
The films, with three different thicknesses between 30 and 150 nm, were prepared by thermally evaporating the material from a Knudsen cell onto a silicon substrate using organic molecular beam deposition (OMBD). The growth rate of about (0.4 ± 0.1) nm min−1 was kept constant with little variation between the materials. The Si(111) substrates were cleaned by keeping them subsequently in acetone, isopropanol, and deionized water in an ultrasonic bath for 5 min each. After that they were dipped for 1 min in 5% hydrofluoric acid in order to remove the native oxide and afterwards once again rinsed in deionized water. This procedure was strictly applied to all metal phthalocyanines but the passivation step (dipping in HF) was omitted in the cases of H2Pc and the porphyrins. For H2Pc only two thicknesses were evaluated.
The ellipsometry measurements were performed with either a VASE or M-2000 ellipsometer, both being variable angle spectroscopic ellipsometers from J A Woollam Co. Inc., Lincoln, NE. The Φ and Δ spectra were measured at the angles 50°, 60°, and 70°. For the data modeling the software WVASE32 and CompleteEASE from the same company were used. The MOKE spectra were measured using a home built spectrometer operating with a photoelastic modulator and lock-in amplifier after [23], as it is commonly used also in reflection anisotropy spectroscopy measurements [24, 25]. The magnetic field during the measurements was 0.35 T (H2Pc, CuPc), 1.05 T (CoPc) and 1.69 T (MnPc, FePc, NiPc, ZnPc, H2TPP, H2TMPP, CuTMPP and NiTMPP). The MOKE spectra were measured in polar geometry, i.e. the magnetic field direction was perpendicular to the sample surface and quasi-parallel to the incident and reflected white light beam. The data shown in this work are normalized to 1 T by scaling the spectra magnitudes linearly with the applied field. This procedure is justifiable, since the strength of the applied magnetic field in our experiment does not influence the lineshape of the spectra.
For the determination of the Voigt constant Q from the measured Kerr angle and from the ellipsometry data the numerical procedure described in [10] was applied. Briefly summarized, the calculations are based on the formula for the complex Kerr angle ΘK
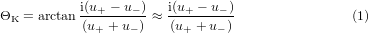
where u± denotes the effective reflection coefficients of the sample for right and left hand circularly polarized light. These can be written as

In the formula the Fresnel reflection coefficient r and the phase propagation term a are appearing. The indices A, F, and S stand for air, film and substrate.

In equation (3)  is the complex refraction index
is the complex refraction index  , ω is the angular frequency of the light, c the vacuum light velocity, and d the thickness of the film. For Voigt constants Q = −iεy/εx much smaller than 1 the modification of the refractive indices for the circular light components can be written as
, ω is the angular frequency of the light, c the vacuum light velocity, and d the thickness of the film. For Voigt constants Q = −iεy/εx much smaller than 1 the modification of the refractive indices for the circular light components can be written as

Thus, with the knowledge of Q, of the film thickness d, and of the ellipsometry result  the complex Kerr angle can be calculated. To go the opposite way one has to invert the function ΘK = f(Q) numerically.
the complex Kerr angle can be calculated. To go the opposite way one has to invert the function ΘK = f(Q) numerically.
For proper modeling of the ellipsometry spectra of the investigated porphyrinoids, uniaxial anisotropy as well as surface roughness has to be taken into account. The main axis of anisotropy is the direction perpendicular to the substrate plane and linear light polarization in that direction experiences a different dielectric function (or optical constants) compared with light with a linear polarization parallel to the sample plane. The origin of the anisotropy stems from a preferential alignment of the molecules on the substrate [10, 26]. For estimating the average tilt angle α of the molecular plane with respect to the substrate plane we assume that all molecules adopt the same orientation and that the transition dipole moments of the electronic transitions in the Q-band (see later discussion) are degenerated and both parallel to the molecular plane. The molecular film is thus considered to consist of perfectly aligned disks. Under these assumptions we can employ the following formula [10]:
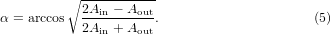
Ain and Aout are the integrals over the area of the Q-band in the spectra of the in-plane and out-of-plane components of the extinction coefficient k, respectively. The surface roughness was modeled by introducing several (typically 11) slices of effective media on top of the Pc film in the model. Bruggeman mixing of the dielectric functions of the Pc and void was applied [27] with different material content in each single slice. The content was varied with an error function (erf), assuming a Gaussian height distribution over the area of the film within the measuring light spot. Therefore on the bottom of the 'roughness slices' the assumed content of Pc material was close to 100% and on the top close to zero. The assumption of a Gaussian profile also allows us to calculate the rms-roughness value. The obtained tilt angles and surface roughnesses will be discussed in section 4.
3. Theoretical methods
3.1. Technical details: DFT calculations
In order to support the experimental results and to further investigate the electronic structure of the materials we performed all-electron DFT calculations of all individual free molecules and for the bulk structures of the MePcs.
The calculations for the individual free molecules were carried out using the NRLMOL program package [28]. NRLMOL combines large Gaussian orbital basis sets, numerically precise variational integration and an analytic solution of Poisson's equation in order to accurately determine the self-consistent potentials, secular matrix, total energies and Hellmann–Feynman–Pulay forces. To include exchange and correlation effects, the generalized gradient functional developed by Perdew, Burke, and Ernzerhof (PBE) was applied [29]. First we relaxed the structures of the individual molecules by performing a full geometry optimization. The symmetries of the initial and the final state are of decisive character for the calculation of the possible optical transitions. The corresponding dipole matrix element is obtained for any possible transition and the intensity of every transition is weighted with the value of the dipole matrix element. This offers a direct mapping of the spectral signals to the electronic structure of the molecule. In order to reach qualitative understanding of our experimental data we added a Gaussian broadening of about 0.15 eV to the theoretical spectra.
For the bulk structure calculations we used the ELK program which is an implementation of the all-electron full potential linearized augmented plane-wave (FP-LAPW) method [30]. The starting structures correspond to the α-phase of MePc and were obtained from the Cambridge Structural Database. The calculations were performed using a basis set with 440 valence states and an additional 562 local orbitals together with the same PBE functional as for the NRLMOL calculations. After geometry relaxation of the structures (Fmax < 0.05 eV Å−1) the full dielectric tensor was calculated according to [31]. The Voigt constant can be retrieved directly from the elements of the dielectric tensor. The effective k-point mesh size used for Brillouin-zone integration in the case of calculation of optical properties was 400. The effect of spin–orbit coupling and a magnetic field normal to the molecular plane were included in the calculations.
3.2. Perimeter model
The basic assumptions within the perimeter model (PM) are as follows.
- (i)The π-system under investigation can be treated independently of the remaining electrons of the molecule.
- (ii)Spin–orbit coupling is negligible small.
- (iii)Full aromaticity of the investigated π-system (i.e. containing (4N + 2) electrons, with
 , with N being a natural number) is assumed, describing a closed path lying approximately in a plane.
, with N being a natural number) is assumed, describing a closed path lying approximately in a plane. - (iv)σπ∗ and nπ∗ state-mixing into π→π∗ transitions will be ignored.
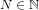
Assumptions (i) and (ii) assure the separability of the molecular wavefunctions (WFs) into three components, one perpendicular to the plane of the molecule z, one polar angle ϕ in the molecular plane and an in-plane radius r. Assumption (iii) validates that outer shell π→π∗ transitions are sufficient for the main part of the optical spectra. Note that the model is no longer valid if the number of electrons in the investigated π-system deviates from the number claimed above. The discussion may then be carried out for a stable aromatic anion or cation instead. Now that we know about the limitations of the PM and can ensure its validity, we will focus on the definition of the orbital angular ring quantum numbers (OARNs).
The wavefunctions predicted by the PM are complex eikϕ with the imaginary unit i, the angular variable ϕ and the OARN k. Commonly the PM is combined with classic molecular orbital theory, which restricts the values of k to  , where n is the number of electrons in the considered π-system. However, one has to note that the selection rules are defined with respect to orbital angular quantum numbers (OANs). If spin–orbit coupling is negligible, the selection rules split up in statements for spin s and angular momentum l resulting in the well known dipole selection rules Δl = ±1, if at the same time spin flips are not allowed. But why is it not possible to state that the OARN is equal to the OAN? Since an arbitrary closed path along a π-system never possesses a C∞-symmetry,
, where n is the number of electrons in the considered π-system. However, one has to note that the selection rules are defined with respect to orbital angular quantum numbers (OANs). If spin–orbit coupling is negligible, the selection rules split up in statements for spin s and angular momentum l resulting in the well known dipole selection rules Δl = ±1, if at the same time spin flips are not allowed. But why is it not possible to state that the OARN is equal to the OAN? Since an arbitrary closed path along a π-system never possesses a C∞-symmetry,  is not a constant of motion. In other words if there is no potential with circular symmetry, the OARN is not equal to the OAN. However, identifying the OARN as the OAN of
is not a constant of motion. In other words if there is no potential with circular symmetry, the OARN is not equal to the OAN. However, identifying the OARN as the OAN of  is often misleadingly done in literature. The interpretation of the OARN as an eigenvalue of the linear momentum of the 1D problem along the circle is invalid, due to the periodic potential resulting from the atoms, along the polar coordinate ϕ. Hence the linear momentum is not a constant of motion in the 1D case.
is often misleadingly done in literature. The interpretation of the OARN as an eigenvalue of the linear momentum of the 1D problem along the circle is invalid, due to the periodic potential resulting from the atoms, along the polar coordinate ϕ. Hence the linear momentum is not a constant of motion in the 1D case.
The molecular WFs are either linear combinations of eigenstates or not eigenstates at all of the angular momentum operator  . That leaves basically three options: (1) direct computation of
. That leaves basically three options: (1) direct computation of  applied on the molecular WF at distinct points, (2) using linear combinations of atomic orbitals (LCAOs) to model the molecular WF at distinct points and obtain the linear combinations of the OAN or (3) using the perimeter model, to gain OARNs k, see [32]. In contrast to the first two, the third option is unique for a distinct WF, even if the WF is not an eigenstate or linear combination of eigenstates of
applied on the molecular WF at distinct points, (2) using linear combinations of atomic orbitals (LCAOs) to model the molecular WF at distinct points and obtain the linear combinations of the OAN or (3) using the perimeter model, to gain OARNs k, see [32]. In contrast to the first two, the third option is unique for a distinct WF, even if the WF is not an eigenstate or linear combination of eigenstates of  . The OARN can be used instead of the OAN, since a one-to-one correlation exists. Thus knowing the OARNs for the initial and final WFs and knowing how OARNs relate to the OANs, we are able to characterize transitions in optical absorption and MOKE spectra. The OARN becomes a measure of the OAN by defining the modulus of the OARN as half the number of nodes with sign change of the WF Ψk(ϕ), along the outermost possible path of the investigated π-system. The sign of the OARN can be defined in two ways. The first possibility would be to identify the sign with the sign of the following equation:
. The OARN can be used instead of the OAN, since a one-to-one correlation exists. Thus knowing the OARNs for the initial and final WFs and knowing how OARNs relate to the OANs, we are able to characterize transitions in optical absorption and MOKE spectra. The OARN becomes a measure of the OAN by defining the modulus of the OARN as half the number of nodes with sign change of the WF Ψk(ϕ), along the outermost possible path of the investigated π-system. The sign of the OARN can be defined in two ways. The first possibility would be to identify the sign with the sign of the following equation:
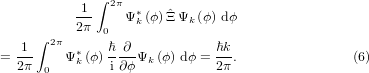
This gives the mean expectation value of the operator  .
.  can be interpreted either as linear momentum operator of the free electron model, i.e. along the circle with sufficient boundary conditions and the variable ϕ, or as
can be interpreted either as linear momentum operator of the free electron model, i.e. along the circle with sufficient boundary conditions and the variable ϕ, or as  of the restrained 3D problem. Note that the 3D problem can be substituted by a 1D problem with coinciding z-axes. Since the radius is fixed, the molecule is approximately planar and the cylinder coordinates are separable.
of the restrained 3D problem. Note that the 3D problem can be substituted by a 1D problem with coinciding z-axes. Since the radius is fixed, the molecule is approximately planar and the cylinder coordinates are separable.
The second way of determining the sign of the OARN is by using classic electrodynamics and Lorentz law. It follows that k < 0 if the magnetic moment, induced by the moving electrons, and the z-axis, are anti-parallel, and k > 0 otherwise. This suggests the illustration of the sign as the circulating sense of the electrons. Thereby the case of maximal k is implicitly defined as positive.
Note that the OARNs k are adding up like scalars to a total ring quantum number K, which follows from the interpretation of  as a linear momentum operator in 1D or from power laws and the form of the molecular WF (a simple product of exponential functions) of n electrons. Therefore it is possible to apply the selection rules on k instead of l, corresponding to a replacement of
as a linear momentum operator in 1D or from power laws and the form of the molecular WF (a simple product of exponential functions) of n electrons. Therefore it is possible to apply the selection rules on k instead of l, corresponding to a replacement of  by
by  , because
, because  on the circle of the perimeter model. The change of the OARN Δk, resulting from a transition, can be recognized as a measure of the circular charge distribution. For example a greater OARN of the excited state can be interpreted as formation of a negatively charged particle. It is now clear that magnetic moments, induced by the circular currents, are proportional as well to Δk.
on the circle of the perimeter model. The change of the OARN Δk, resulting from a transition, can be recognized as a measure of the circular charge distribution. For example a greater OARN of the excited state can be interpreted as formation of a negatively charged particle. It is now clear that magnetic moments, induced by the circular currents, are proportional as well to Δk.
In order to assign OARN one can start from the geometry of the molecule with the corresponding real-valued representations of the WF of interest and condense it using the following three steps.
- (i)Determine whether the molecule in question is aromatic or not. If it is not, use any stable aromatic anion or cation derivable from the molecule.
- (ii)Determine the number of electrons in the π-system under investigation.
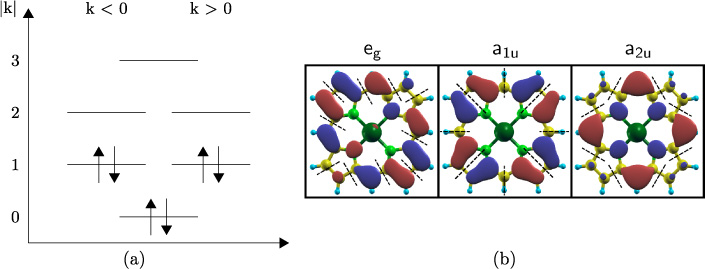
- (iii)Plot the real-valued WFs and count the number of nodes with sign change along the path of the π-system of interest, this would equal figure 1. Now half the number of counted nodes equals the number of nodal planes, i.e. the absolute value of the OARN k.
Finally we are going to remark on possible influences of the center atoms, which will support our discussion and can be understood with the help of the WF symmetry. All metal atoms have lower electronegativities than hydrogen and therefore the binding σ-electrons will be shifted towards the ring. This will increase the electron densities on the nitrogen sites and increase the energies of states with electron density there by Coulomb interaction. This effect is known as the inductive effect.

Figure 1. For a (14)-annulene (phenanthrene; left), the 'unfolded' molecule (middle) and model perimeter (right) with atoms and nodal planes highlighted are shown. The two degenerate sets of nodal planes of every plot are shown for k = 2. (Analogous to [14].)
Download figure:
Standard image High-resolution imageThe next effect is known as the conjugative effect and influences primarily the π-system of the TMPPs. As the p orbitals, especially pz, of the d-block elements are empty, transition group elements increase the size of the π-system by conjugation. The energy of a WF with according symmetry, i.e. without nodal points on the center atom, is lowered.
4. Results and discussion
4.1. Optical properties: B- and Q-band
The B-band was defined as the energetically higher feature rising at ∼2.8 eV for TMPPs in the optical spectrum based on an a1u→eg transition. For porphyrins, experiments indicate, in agreement with theory, the B-band being related to π→π∗ transitions of the aromatic system, nearly independent of the center atom(s) (see [33, 22]). Also the methoxy sidegroups do not influence the lineshape of the B-band significantly.
The Q-band is lower in energy at ∼2.25 eV for TMPPs and based on an a2u→eg transition. It was also found that the Q-band is dependent on the central atom(s), since a WF of a2u symmetry has no nodal point at this location. Figures 2 and 5 show this dependence by comparing H2- and the MeTMPPs. The former shows a four-fold splitting of the Q-band, since the total spin is 0 and the unoccupied states are approximately degenerated within 10 meV. This can be related to the C2 symmetry of the molecule. Please note that the definition of Q- and B-band by using the energy can be misleading, as in the case of Pcs the character of the two features exchanges with the a2u→eg transition occurring at higher energy in that case.
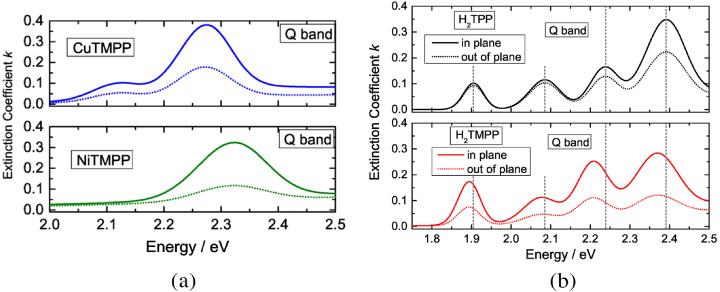
Figure 2. Measured extinction coefficients k of the Q-band. (a) Typical lineshapes for MeTMPP for Cu ( ) and Ni (S = 0). (b) Effect of introducing methoxy sidegroups into TPPs. The slight redshift of the H2TMPP films may indicate a higher degree of crystallinity of H2TMPP.
) and Ni (S = 0). (b) Effect of introducing methoxy sidegroups into TPPs. The slight redshift of the H2TMPP films may indicate a higher degree of crystallinity of H2TMPP.
Download figure:
Standard image High-resolution imageThe influence of the methoxy sidegroups on the shape of the Q-band is shown in figure 2(b). One can see a slight redshift of the functionalized molecule and a change in the ratio between in-plane and out-of-plane components of the extinction coefficient. The redshift of the H2TMPP absorption is most probably an effect of stronger intermolecular interactions, an effect known as crystallochromy, which might be an indication of a better degree of crystallinity of these films.
As can be seen from DFT calculations, each of the a1u→eg and a2u→eg transitions ends in the same final pair of states with eg symmetry. This is true for all investigated molecules, see figure 3. The reversal of Q- and B-band ordering between TMPP and Pc is related to a change in level ordering of the corresponding occupied states. Accordingly the a1u orbital is once above (TMPP) and once below (Pc) the a2u orbital, as was already observed in the case of manganese derivates [34]. These four states account for the Q- and B-bands of porphyrins [35] and the description is known as Gouterman's four orbital model [36], which was used successfully for various porphyrinoids [19, 22, 33, 36–38].
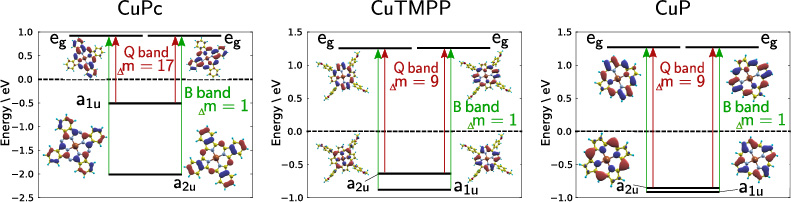
Figure 3. Molecular orbital scheme of selected orbitals of the ground state for CuPc, CuTMPP and as reference CuP with indicated Q- and B-band transitions.
Download figure:
Standard image High-resolution imageIn figure 4(b) it can be seen that the Q-band of all Pcs consists of at least two main features. H2-, Co-, Ni-, Cu-, and ZnPc all show significant splitting of the Q-band into main features at ∼1.75 and ∼2.1 eV. The Q-bands of Mn- and FePc show an even more complex character and in contrast to the other Pcs significant absorption at low energies (<1.5 eV ) is observed. The structure of the MePc Q-band (1.5−2.5 eV ) has been studied using different experimental and theoretical techniques, but there is so far no agreement as to what are the main effects that determine the observed line shapes. The proposed models discuss intramolecular excitation, vibronic coupling, lifting of degeneracy due to lowered symmetries in a crystal, or Davydov splitting (see e.g. [39] and references there). However, none of the approaches is able to explain the experimental spectra quantitatively, and even the qualitative agreement between experiment and theory is often unsatisfactory. Knupfer [39] and Wojdyla [40] therefore proposed the inclusion of charge transfer excitations which can explain the splitting of the Q-band in CuPc very well. The inclusion of different charged species to explain the experimental optical data of MnPc was also discussed before [41]. Here, we will extend this idea to all Pcs.
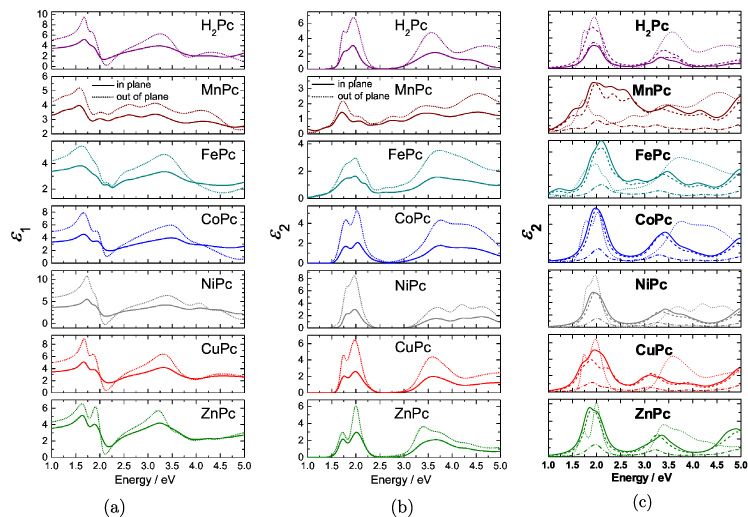
Figure 4. Optical constants of the Pcs under investigation. (a) Real parts and (b) imaginary parts of the dielectric tensor. Solid lines are the in-plane and dotted lines are the out-of-plane components. (c) Comparison of the DFT calculated imaginary parts εxx (dash-dotted), εyy (solid) and εzz (dashed) of the dielectric tensor compared with the measured out-of-plane component (dotted).
Download figure:
Standard image High-resolution image
Figure 5. Optical constants of the TMPPs under investigation. (a) Real parts and (b) imaginary parts of the dielectric tensor. Solid lines are the in-plane and dotted lines are the out-of-plane components.
Download figure:
Standard image High-resolution imageIn figure 4(c), we show our DFT results of ε2 for the different Pcs. The bandgap of standard DFT calculations is systematically too small [42, 43], and therefore the calculated optical spectra are rigidly shifted by ∼0.5 eV. The overall agreement of the theoretical data with the measurements is rather good. The most apparent difference, however, is the lack of the Q-band splitting for Co-, Ni-, Cu- and ZnPc in the calculations.
To understand this major difference we estimated the size of all previously proposed effects on free molecules. We found for example that breaking the symmetry of the molecules (Davydov splitting) cannot explain the magnitude and the direction (i.e. shifts to higher instead of lower energies) of the Q-band splitting of the Pcs. Following the idea of charged species [41] we performed calculations on doped molecules. Our results are shown in an exemplary manner for ZnPc in figure 6 and are similar for all investigated Pcs. With reduction of the electron count, we see a broadening and splitting of the Q-band. At the same time a shift of ∼0.3 eV to lower energies can be observed, which is a perfect match to experiment. Combining spectra from doped and undoped ZnPc with different weights can easily reproduce the experimental lineshape. Thus we conclude that the main effect of the observed Q-band splitting has its origin in the existence of charged species. The question about the exact origin of the charged species needs to be addressed in further investigations. It is possible that the charged species arise during the excitation experiment, that thin-film defects lead to intrinsic doping of the material or that the observed charged species arise from the coupling of charge transfer states with Frenkel excitons, analogous to the situation found in perylene pigments [44].
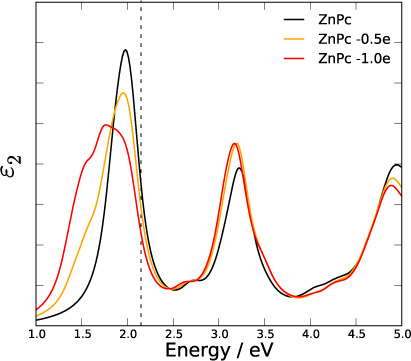
Figure 6. Calculated ε2 for different effective doping levels of ZnPc using 0.5 and 1.0 electrons fewer per molecule with 2 molecules per unit cell.
Download figure:
Standard image High-resolution image4.2. Determination of average tilt angle
As discussed earlier (see equation (5)) the optical spectra allow for an estimation of average tilt angles by using the in-plane Ain and out-of-plane Aout components of the extinction coefficient k. For H2Pc, CoPc, NiPc and CuPc, Aout is significantly larger than Ain as shown in figure 4. This indicates a standing configuration of the molecules on the substrate (molecular plane close to perpendicular to the substrate plane). For MnPc, FePc, and ZnPc, Aout is only slightly larger than Ain, which points to a slight preference for crystallites with standing molecules, i.e. average molecular tilt angles between 60° and 75°.
In the case of the investigated TMPPs Ain is higher than Aout. The estimated average tilt angles of porphyrins range between 35° and 47°. Figure 2(b) shows a higher ratio between the Ain and Aout of H2TMPP compared with H2TPP. This indicates, together with the crystallochromy, that the presence of methoxy sidegroups seems to introduce a better ordering of the molecules. The surface roughness values are obtained as stated in the experimental section. In the case of Pcs they range from (2.5 ± 1.0)% of the total film thickness for H2Pc and MnPc to (9.5 ± 1.5)% for CuPc. For PP films, the roughness is in the range of 1–5% of the film thickness.
4.3. Magneto-optical properties: Voigt constant
For the Pcs the spectral dominance of the Q-band is even more pronounced in the Voigt constant Q than in the extinction coefficient k. This is a bit surprising because both Q-band (∼2 eV) and B-band (∼3.5 eV) are supposed to absorb into the eg LUMO of the metal Pcs (or into two states arising from the LUMO splitting, b2g and b3g, in the symmetry reduced H2Pc [45]). Transitions from or into degenerated orbitals are responsible for features in magneto-optical spectra [11, 46]. The degeneracy of the excited state is lifted in the magnetic field due to the Zeeman effect [11]. The magnitude of the Q-band feature therefore depends on the orientation of the xy-degenerate band with respect to the incident light. If the molecular plane is parallel to the light polarization, the light can most effectively couple with the dipoles. Therefore in the polar MOKE geometry (analogous to the Faraday geometry) a lying molecule exhibits the largest magneto-optical response and the magnitude of the Voigt constant will scale with the cosine of the molecular tilt angle. This explains the larger magnitude of the signal of the porphyrins which were already found by ellipsometry to align more parallel to the substrate.
Figures 9(a) and (b) summarize the results of the DFT calculations on single molecules of Pcs and TMPPs, yielding to the same values of total spins for identical central atoms. The chemical environment of the central atom is the same in both cases and all differences between TMPPs and Pcs can be accounted for by perturbations resulting from the substituents. This makes these two classes perfect for analyzing the influence of different substituents. However, the molecular spin appears to have either no, or only a small, influence on the value of Q, as the comparison between figure 4 and the single molecule spin-states in figure 9(a) shows. A definite statement regarding this is difficult as the influence of the tilt angles seems to be too large.

Figure 7. Measured Voigt constant dispersions ((a) real part, (b) imaginary part) of the Pcs under investigation. The magnitude is normalized to the expected value at 1 T.
Download figure:
Standard image High-resolution image
Figure 8. Measured Voigt constant dispersions ((a) real part, (b) imaginary part) of the TMPPs under investigation. The magnitude is normalized to the expected value at 1 T.
Download figure:
Standard image High-resolution image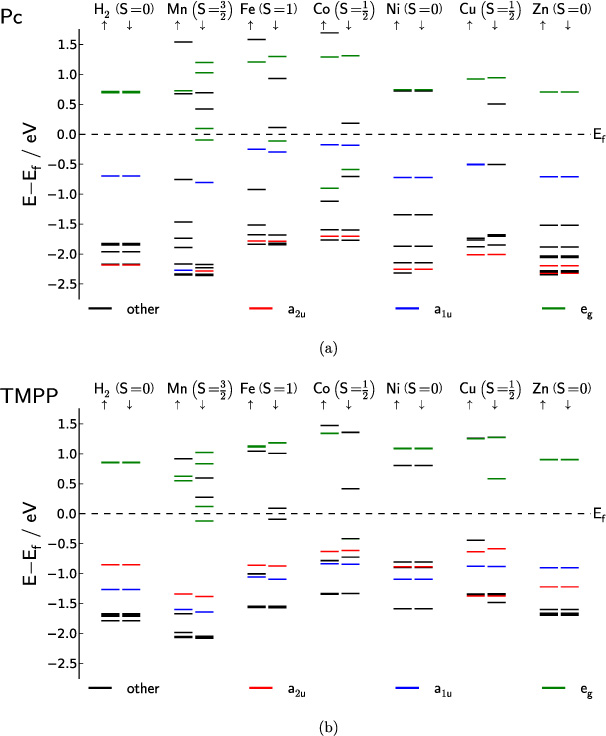
Figure 9. Calculated energy levels aligned with the Fermi level (Ef) of the isolated free molecules of Pcs (a) and the respective TMPPs (b) using the NRLMOL code. States involved in the Q- and B-band transitions are highlighted. For Mn-, FePc the eg-states below Ef are highlighted because they take part in transitions of eg→a2u character with unoccupied final state. For CoPc a transition of eg→a2u character with empty final state can be seen.
Download figure:
Standard image High-resolution imageFor the presented molecules the calculated Voigt constant dispersion was very similar between the samples of different film thickness. Therefore only one representative spectrum of the real and the imaginary part for each material is shown in figures 7 and 8.
The trend of increasing magnitude with decreasing tilt angles found for the ellipsometry data does not match the magnitude variations of the Voigt constant of the Pcs (figure 7). If solely the effect of the molecular orientation on the magnitude of the Voigt constant is considered, the magnitude should be smallest for CuPc and largest for FePc, which is not the case. Another effect influencing the magnitude of the Voigt constant is inter-molecular interaction in the film. This can induce slight deviations from the single molecule symmetry, which can be the reason for the smaller Q-band feature of MnPc in the Voigt constant. In the literature, hints for the symmetry reduction in the case of MnPc are given, although they were previously dismissed as computational artifacts [11, 47]. Another reason for the small Voigt constants of MnPc and FePc is developed in [48], namely that multiple electronic configurations are competing in a very sensitive way with regard to the chemical environment.
The effect of the center atom(s) on the Q- and B-band of TMPP is essential for the understanding of Porphyrinoid spectra. For MePPs with D4h symmetry it was found [33], that there is a linear correlation of electronegativity and transition energy of the Q-band. As the electronegativity drops the transition energy increases. This correlation results from inductive and conjugative effects. The inductive effect increases the orbital energies of eg and a2u if the electronegativity of the center atom(s) drops. The conjugative effect, on the other hand, lowers the energy of only a2u if electronegativity is lowered, as a1u and eg have nodal points on the central atom site.
4.4. π→π∗-transitions in the perimeter model
Now we will discuss the application of the perimeter model to describe the π→π∗-transitions of TMPPs and Pcs. Following the algorithm of the theoretical section, we have to check for aromaticity and determine the electron numbers. There are 16 atoms in the circumference of the TMPPs (hydrogen is always omitted) and therefore an adequate perimeter model would be based on the (16)-annulene di-anion, [C16 H16]2−. For TMPPs we obtain the same result neglecting the substituents, which can be done as they do not interfere with the π-system. The chosen anion has then in total 18 electrons in the π-system, which agrees with values obtained for TPP in literature [16, 19, 38]. As only the main π-system is of interest the values for TPP and TMPP coincide.
The same procedure gives 36 atoms for Pc and therefore the PM should be based either on the (36)-annulene di-anion, [C36 H36]2−, or on the (36)-annulene di-cation, [C36 H36]2+. Michl [16] has shown that the total number of electrons in the π-system should be 34. Hence the latter choice is the best approximation, although a di-cation of Pc may not be existent in nature. Using Gouterman's 4-orbital model [36], the perimeter model has to be applied to only four different WFs of correct symmetries with energies near the Fermi level.
This gives, according to figure 10(b), the following absolute values of the OARN for the investigated porphyrinoids [21]: Ψ1, Ψ2 have k = 4 and Φ1, Φ2 have k = 5 for the TMPP. For the Pc it is k = 8 and k = 9, respectively. The sign has to be chosen by following experimental findings, since the WFs, like in figure 10(b), are linear combinations of positive as well as negative OARNs.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 10. (a) Molecular orbital scheme of the ground state of the perimeter model for benzene with OARN k. The degenerated levels of the ground state are occupied according to Pauli's principle. (b) Real-valued linear combinations of WFs (red = positive; dark blue = negative) from CuP, which is symmetry-equivalent to CuTMPP (figure 3) (dark green = Cu; light green = N; yellow = C; light blue = H) with the same symmetry but differing sign of k. Cases shown are eg with |k| = 5, a1u and a2u both with |k| = 4. Nodal planes are indicated by dashed lines.
Download figure:
Standard image High-resolution image{kind=link}
As we can see in figure 5 the B-band is a very strong transition for TMPPs at higher energies than the Q-band. Thus the B-band has to be a transition between molecular WFs of the same sign, i.e. ± 4→ ± 5. This is the only choice possible, which is dipole allowed according to the selection rule Δk = ±1. The Q-band will be weakly forbidden as it will correspond to transitions ± 4→ ∓ 5, yielding Δk = ±9. The distorted character of the porphyrinoids with respect to the perimeter model is obvious as the Q-band does not vanish completely. The assumption of dipole transitions is not crucial here, because even quadrupole moments are not sufficient for Δk = ±9. A symmetry classification and interpretation of DFT data confirmed that the stronger transition at higher energies is of a1u→eg character for the TMPPs. Thus the a2u→eg electronic transition will be weakly symmetry forbidden for TMPPs (Q-band) and Pcs (B-band).
In figure 5 one can see that this holds for all TMPPs investigated. For Pcs (figure 4) however, the forbidden transitions seem to be allowed nearly completely. If the perimeter model would hold strictly, then all molecular orbitals belonging to the same OARNs would be degenerated, see figure 10(a). As discussed the final states of the main electronic transitions of Q- and B-bands are the same and of equal OARN. Since a1u and a2u also share the same OARN, they would have to be degenerated for Pcs as well as for TMPPs. In fact the degeneracy is always lifted because the porphyrinoids can be represented only by perturbed perimeters. Therefore we can take the energy difference of both bands as an indicator of the validity of the perimeter model. Since figures 4(b) and 5(b) show clearly that the energy difference is much larger for the Pcs (e.g. NiPc with ∼2.25 eV; NiTMPP with ∼0.5 eV), the perimeter model is assumed to hold less strictly. In other words, the selection rules based on the OARNs are weakened and intensities of the Q-band should be more alike for the different Pcs.
5. Summary
We presented an experimental and theoretical comparison of optical and magneto-optical properties of metal phthalocyanine and methoxy functionalized metal tetraphenylporphyrin thin films prepared on silicon substrates. These materials can easily be functionalized and the magnetic properties engineered by changing the central metal atom or adding substituents, which makes them promising candidates for possible application in molecular electronics and spintronics. In particular the materials investigated and discussed are MePc with Me = Fe, Co, Ni, Cu, Zn and MeTMPP with Me = Cu and Ni. We also compared our results to the metal-free H2Pc, H2TPP and H2TMPP.
Using density functional theory calculations augmented by the perimeter model the observed spectra can be understood and differences between the TMPPs and Pcs rationalized. The simple perimeter model allows for qualitative understanding of optical transitions in planar, aromatic molecules and is able to qualify differences between Pc and TMPP type materials. Using DFT together with the simple perimeter model appears to be a very powerful and general method to analyze planar aromatic systems.
One of our main findings using the perimeter model is the change in symmetry of the transitions involving the Q- and B-band between the Pcs and TMPPs for all members of the series investigated. Furthermore we could demonstrate that the perimeter model works well in the case of TMPPs. However it has a limited applicability in the case of Pcs because the a1u and a2u states would be degenerated in the model, in contrast to reality.
The reason for the splitting of the Q-band for the Pcs has been discussed quite extensively in the past. The proposals include intramolecular excitation, vibronic coupling, lifting of degeneracy due to lowered symmetries in a crystal or Davydov splitting (see e.g. [39] and references there). Following earlier discussions for CuPc [39, 40] and MnPc [41], we extend the idea of charge transfer states as a reason for the splitting of the Q-band to all MePcs. This allows us to understand the measured optical spectra of the whole series rather well. Our results further indicate that symmetry lowering and lifting of degeneracies are most likely not sufficient to explain experimental observations. At the same time the detailed mechanism of the charge transfer states is not clear and requires further work.
We hope that our work will stimulate further research on these molecular materials in order to elucidate the potential of the Pcs and TMPPs for molecular electronic and spintronic application.
Acknowledgments
The authors would like to thank the DFG research group FOR 1154 Towards molecular spintronics for financial support and the ZIH TU-Dresden for providing computer resources, technical expertise and assistance. C Martin would like to thank the SAB for support by an ESF grant (ESF:080945373). We thank C Röder for technical assistance.
Footnotes
- 3
Porphyrinoid refers to compounds with the porphyrin skeleton, i.e. porphyrins (Ps), meso-tetra-kis-(3-methoxy-phenyl)porphyrin and phthalocyanines are included besides others.