
Abstract
Molecular dynamics is an active area of research, focusing on revealing fundamental information on molecular structures and photon–molecule interaction and with broad impacts in chemical and biological sciences. Experimental investigation of molecular dynamics has been advanced by the development of new light sources and techniques, deepening our understanding of natural processes and enabling possible control and modification of chemical and biomolecular processes. Free-electron lasers (FELs) deliver unprecedented intense and short photon pulses in the vacuum ultraviolet and x-ray spectral ranges, opening a new era for the study of electronic and nuclear dynamics in molecules. This review focuses on recent molecular dynamics investigations using FELs. We present recent work concerning dynamics of molecular interaction with FELs using an intrinsic clock within a single x-ray pulse as well as using an external clock in a pump–probe scheme. We review the latest developments on correlated and coincident spectroscopy in FEL-based research and recent results revealing photo-induced interaction dynamics using these techniques. We also describe new instrumentations to conduct x-ray pump–x-ray probe experiments with spectroscopy and imaging detectors.
Export citation and abstract BibTeX RIS
1. Introduction
The motion of electrons and atomic nuclei in molecules is fundamental to all physical and biological systems. Knowledge regarding the electronic and nuclear motions underpins our understanding of chemical reactions and biological processes [1]. Despite tremendous experimental and theoretical efforts, the central questions of how molecular structures change and how energy flows through all transient stages from the initial to the final products are, in most cases, still not fully understood. Nuclear motion occurs on femtosecond time scales and Angstrom spatial scales. Electron motion is often much faster, particularly for tightly bound inner-valence and core electrons. New tools developed in the past two decades have made it possible to develop probes that can match many of these spatio-temporal requirements. Ultrafast infrared and optical wavelength lasers have allowed to follow the vibrational, rotational and dissociative motion of molecules [2, 3], and have also led to some degree of quantum control of valence electrons. The more recent advancements in attosecond lasers now promise some direct control of the electronic motion as well [4]. Ultrafast soft x-rays from free-electron lasers (FELs) have photon energies sufficient to access core- and inner-shell electrons, and their femtosecond pulse durations and high intensities provide a powerful tool for time-resolved studies of molecular structural dynamics. The element-specificity of soft x-ray absorption has now been used to chart photochemical reactions and bioprocesses with atomic spatial and femtosecond temporal resolution [5–11]. These advances may soon be able to answer the key questions about energy flow and transient processes in molecular dynamics. We present in this article a compilation of FEL-based investigations that have contributed to improving our understanding of molecular dynamics.
1.1. Pump–probe schemes to take snapshots of dynamics
Complementing the development of ultrafast lasers, pump–probe experimental techniques have been developed, in which a 'pump' laser initiates dynamics which are then captured by a 'probe' laser operating in delayed coincidence. The broadband optical coherence needed to produce ultrafast laser pulses can also excite a coherent superposition of multiple electronic states, thereby forming electronic wavepackets. The evolution of wavepackets can be probed to reveal the dynamics of the photoexcited system under study [12], or map out structural information about the system such as the position of potential energy surfaces [13, 14]. The ultrashort laser-pulse durations allow us to accurately define the initiation time and the time for probing to build a dynamical picture of the evolution of a system on the femtosecond time scale.
1.1.1. FELs as a pump or a probe
Soft x-rays interact most strongly with highly localized core electrons near their photoemission threshold, and this gives x-ray FEL absorption experiments a localized excitation specificity far smaller than the wavelength of the light. This site specificity is a useful feature in many kinds of soft x-ray spectroscopy, including x-ray absorption, photoemission, Auger emission, and x-ray fluorescence [15]. Chemical dynamics and reactions initiated or probed in this way can be studied with atomic spatial resolution: We can interrogate a selected atom in a molecule, and therefore, isolate the dynamic processes at specific sites. Inner shell interaction also produces hole-states which relax via Auger decay processes or fluorescence. Both the hole-state and the transient state following the relaxation processes are highly excited and generally not accessible with optical lasers. Hence, using an x-ray laser as a pump, we are able to study dynamics involving deeper-shell electrons. Hard x-ray FELs with photon energies beyond the binding energies of the light elements can also probe structure through simple diffractive elastic scattering, once their wavelengths are comparable with or shorter than the interatomic spacing or structural feature of interest [16, 17].
In general, pump–probe gas phase spectroscopy is intrinsically nonlinear because it depends on a reasonable probability that the same molecule will interact with both the pump and the probe laser. Therefore these methods are only generally useful when the total probability for absorption or scattering is on the order of 10% per molecule. For typical megabarn cross sections and 100 fs pulses, this implies a focused x-ray intensity exceeding 1014 W cm−2. In experiments with optical lasers, this is well into the strong-field regime where perturbation theory breaks down and ponderomotive forces control the dynamics. Fortunately, however, the ponderomotive potential scales as λ2, so at kilovolt photon energies the ponderomotive effects are reduced by a factor of about a million. Thus x-rays of short duration and high intensity have become a versatile probe for investigations of molecular dynamics.
1.1.2. Clock in pump–probe schemes to time the snapshots of the dynamics
To reveal the dynamics, we need to take snapshots of the systems at different times, so good synchronization between the pump and probe pulses is a crucial element in pump–probe experiments. The simplest clock is the laser carrier frequency itself, and this is used in strong-field experiments using infrared lasers. In these cases the dynamical processes of ionization and recombination or rescattering can be controlled by the laser field during a single cycle. Multiple path interference can be observed when an ionized electron wavepacket returns to and interferes with the ion core. This recollision process can be tuned by changing simple experimental parameters such as the ionizing pulse wavelength [18, 19]. Multiphoton ionization can also be used as an intrinsic clock in recording the dynamics of the photoionization and molecular fragmentation processes—each photoionization lifts the molecular ions to a new potential energy surface and the sequential photoionization functions as a multiple pump–probe approach where the average delay between every two photoabsorptions is determined and can be tuned by the ionizing pulse intensity [20]. Different processes occurring on the same system may have different time scales which can be exploited as start-and-stop watch in probing molecular structural changes. For example, x-ray emission is a relatively slow process compared to photoionization which is considered nearly instantaneous. Comparison between photoelectron and fluorescence spectroscopy gives the information about the bond length change of the relaxing state on the time scale of the states lifetime [21]. The core-decay mechanics have been used explicitly as an internal clock, referred to as core-hole-clock, to study the electron dynamics in condensed phase in the range of femtoseconds down to hundred attoseconds [22, 23].
The clock for dynamic snapshots can also be external, using a second laser pulse as the probe as has been applied in typical pump–probe experimental settings. The delay time between the pump and the probe pulse can be systematically controlled. Optical-x-ray-FEL pump–probe experiments have been carried out, where the optical lasers may pump the sample to the desired states initiating valence- or inner-valence dynamics and the FEL probes the motion of interest [24, 25]. In this type of experiments, time-jitter between the two independent laser pulses has been reduced in post-experimental data analysis using the information from optical–x-ray cross correlators [25–27]. X-ray/XUV-pump–x-ray/XUV-probe experiments have been realized with the recent development of a split-and-delay apparatus [28–30], providing a jitter-free pump–probe tool for studies of molecular dynamics.
1.2. Measurement methods: coincidence and covariance techniques to track molecular fragmentation
Various pathways in molecular interaction with photons and subsequent molecular dissociation may lead to the same products. Disentangling these different pathways and the corresponding mechanisms has presented challenges. Correlation methods have been implemented, such as coincidence and covariance measurements and analysis methods, and have produced promising results [31–35]. Correlation measurements involving different spectroscopies and experimental approaches, such as electron-ion [36], ion-x-ray-emission [37], x-ray-emission-x-ray-diffraction [38] correlations, provide multiple views of the system under study and serve as cross-checks, to validate interpretations of the underlying dynamics. Multi-dimensional correlations can in principle resolve multiple bond cleavages, or show the presence of entangled fragmentation pathways [39, 40]. These techniques applied to pump–probe studies can contribute to deeper understanding of the molecular dynamics, especially when combined with spectral or spatial information.
2. Probing molecular dynamics using multiphoton ionization as an intrinsic clock
2.1. Introduction
We have investigated the fragmentation dynamics of simple diatomic N2 molecules using multiphoton ionization. Specifically, we used the multiple photoionization (P) and Auger (A) cycles as an intrinsic clock to probe the development of the ionization and dissociation of N2. In other words, we can regard the multiple PA cycles by sequential absorption within one single x-ray pulse as a pump-multiple probe scheme. This scheme should be applicable for any system [20].
The high x-ray FEL intensity with photon energies above the binding energies of core electron in neutral molecules, as well as molecular and atomic ions, enables sequential multiphoton absorption at core-shell levels [41]. Following core-shell ionization, the resultant ionic states are highly exited and relax mainly through Auger decay processes in light elements. At the current available Linac Coherent Light Source (LCLS) pulse intensity, the dominant scenario of the interaction is that each photoionization is followed by Auger decay processes before the next photoabsorption. This PA sequence repeats until too few electrons remain to allow the Auger decay process. The molecular ions may continue absorbing multiple x-ray photons sequentially (PP sequence) until all the electrons are stripped off. These PAPA... sequences followed by PP sequences take place while the molecule ions dissociate within the time scale of a single x-ray pulse which is up to a few hundred femtoseconds [20, 41, 42]. For a particular molecule, how fast these sequences proceed is basically determined by the photoionization rate, which can be controlled by varying the ionizing beam parameters. In other words, beam parameters such as intensity can be used as a control knob to modify the ultrafast dynamics in the interaction between molecules and x-ray pulses.
The investigation of N2 dynamics via multiphoton ionization represents a showcase of molecular dynamics study using ultrafast and intense FEL pulses applicable to any molecule and FEL source. At the first PA step, the main relaxation channel leaves the N2 molecule doubly ionized, initiating molecular dissociation. Each further PA step populates the dissociating molecular ions to a new ionic state of higher charge associated with a different asymptotic dissociation limit, described as N Nn + +Nm + or (n,m) channel. The kinetic energy (KE) of atomic ions of each final charge state results from the accumulation of KE gained through all ionization intermediate steps, hence is determined by the internuclear separation at the time of photoionization and the corresponding potential curves. With the measurement of the final KE of the fragment ions and a simple numerical model, we mapped the photoabsorption step in the time and the internuclear separation domains, i.e., to time-resolve the dynamics of the multiphoton ionization process. We observed the modification of the dynamics by varying the beam pulse duration at the same pulse energy, as well as the different dynamics at different beam intensities in the different spatial regions of the x-ray beam [20].
Nn + +Nm + or (n,m) channel. The kinetic energy (KE) of atomic ions of each final charge state results from the accumulation of KE gained through all ionization intermediate steps, hence is determined by the internuclear separation at the time of photoionization and the corresponding potential curves. With the measurement of the final KE of the fragment ions and a simple numerical model, we mapped the photoabsorption step in the time and the internuclear separation domains, i.e., to time-resolve the dynamics of the multiphoton ionization process. We observed the modification of the dynamics by varying the beam pulse duration at the same pulse energy, as well as the different dynamics at different beam intensities in the different spatial regions of the x-ray beam [20].
2.2. Experimental and analysis method
2.2.1. Experimental setup
The experiment was carried out at the AMO end-station at LCLS) at Stanford Linear Accelerator Center National Laboratory (SLAC). We used the high field physics chamber which is equipped with an ion time of flight spectrometer (iTOF) and five electron time of flight spectrometers . The work presented here shows only the iTOF data. The iTOF axis is perpendicular to both the polarization and propagation direction of the x-ray beam. N2 molecules were delivered to the interaction region by a horizontally oriented super sonic jet with 100 μm pulsed nozzle which is positioned parallel to the direction of the beam polarization. The gas density of the molecules in the interaction region was estimated to be ∼1010 particles per cm3. The photon energy is 1.1 keV (±15 eV). The pulse energy was measured upstream of the beam transport and was reduced by 65%–85% at the interaction region due to the optics losses. The x-ray beam was focused to an area of about 2 μm2 by Kirkpatrick–Baez (KB) mirrors. For details of the experimental setup, see [7, 41].
2.2.2. Extraction of fragment KE
For different narrow windows of initial ion KE, we simulated using SIMION the iTOF spectra which have Gaussian-profiles and are used as base functions. We extracted the KE distribution for each atomic charge state by fitting the measured spectral peak with the base functions by varying the heights of the base functions. The simulation, hence, the fitting procedures include the spectrometer transmission. We set an isotropic spatial distribution and a marrow Gaussian KE distribution for initial ions. Figure 1 shows an example of the obtained final KE distribution for different charge states at pulse duration 110 fs. The KE distribution changes slightly depending on the initial values for the fitting parameters. However, the average KEs obtained from the distribution are robust and do not depend on the initial fitting conditions [20, 43].

Figure 1. Kinetic energy distribution for different charge states at x-ray pulse duration of 110 fs. The distribution was obtained by fitting the TOF spectra with base functions generated by SIMION (see text for details).
Download figure:
Standard image High-resolution image2.2.3. Model to obtain the timing information of the dynamics
With the knowledge of the potential curves and ionization rates, we built a simple numerical model to obtain the timing information of the multiphoton ionization with the fragments' KEs. For particular fragmentation pathways, we generate curves of average time interval between photoionization as a function of KE for different final atomic charge states. In this model, we adapted a few assumptions. (1) We assumed, based on experimental evidences, a dominant fragmentation pathway, which is N2 →(1,1)→(3,1)→(3,3)→(3,5)→(5,5)→(6,5)→(6,6)→(6,7)→(7,7). (2) We approximated the potential curves for high charge sates with Coulomb potentials. (3) We assumed the Auger decay process is instantatenous, i.e., the molecular ions do not have time to dissociate during the Auger decay processes. (4) We set the time interval between photoabsorption inversely proportional to the cross section of the atomic ionization. Using this model and with the measured fragment KE, we obtained the average time interval between photoionization steps, as well as the corresponding internuclear separation for the photoionization [20].
2.3. Experimental results and discussion
Figure 2 shows the TOF spectra of N2 measured at 1.1 keV photon energy, similar pulse energy and three different pulse durations, 80, 110 and 280 fs. We observed fully stripped atomic N ions as a result of the PAPA...PP sequences. As shown in figure 2, the spectral width changes as a function of pulse duration, increasing for shorter pulses. To quantitatively show the KE dependence on pulse duration, we extracted the average KE for the atomic ions, as has been described in 2.2.2. Figure 3 shows the mean KE for all seven different charge states measured at the three pulse durations. As seen in figure 3, the KE increases for increasing charges, a charge-state dependence shown for all three pulse durations. For a particular charge states, higher mean KEs are associated with shorter pulse durations, i.e., higher pulse intensities—a pulse-duration dependence observed at charge states higher than 2+. For N+ atomic ions, the photoionization occurs at the equilibrium position of neutral N2, independent of the pulse duration. This timing is taken as the common starting time for the multiphoton ionization process at different pulse durations and is different from the absolute time along the temporal profile of the x-ray pulse. Using the potential curves for dissociating N , the calculated KE for dissociation of the (1,1) channel initiated at the equilibrium internuclear position is consistent with our measurement. The main contribution to the (2,2) channel is supposed to be dissociation of N
, the calculated KE for dissociation of the (1,1) channel initiated at the equilibrium internuclear position is consistent with our measurement. The main contribution to the (2,2) channel is supposed to be dissociation of N following the PA process in the semibound N
following the PA process in the semibound N ionic states, consequently leading to KEs that are not affected by the pulse duration due to the absence of dissociation.
ionic states, consequently leading to KEs that are not affected by the pulse duration due to the absence of dissociation.
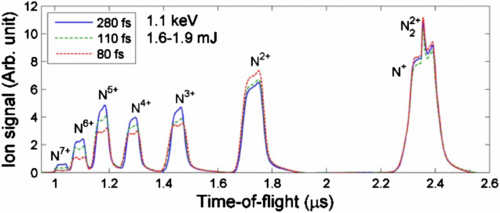
Figure 2. ITOF spectra of N2 at three different pulse durations (80, 110 and 280 fs). Data were taken at photon energy 1.1 keV and pulse energy 1.6–1.9 mJ per pulse. Each spectrum was normalized to the total ion yield at the corresponding pulse duration.
Download figure:
Standard image High-resolution image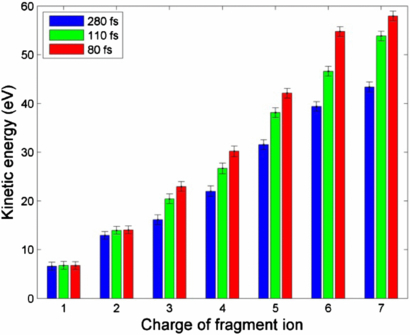
Figure 3. Average KE for different atomic charge states measured at three different pulse durations, extracted from the TOF data shown in figure 2, where data were taken at photon energy 1.1 keV and pulse energy 1.6–1.9 mJ per pulse. Reprinted with permission from [20]. Copyright 2012 by the American Physical Society.
Download figure:
Standard image High-resolution imageWe obtained the average time interval and the internuclear separation position for each photoionization event for different fragmentation pathways at the three different pulse durations (see figure 4). We examine the results in two ways. First, as shown in figure 4(a), we compare the dynamics at different pulse durations for a fragmentation pathway associated with a particular final charge state which is 7+ in figure 4(a). At shorter pulses, the average time interval between photoabsorption is smaller than at longer pulses, and correspondingly the photoionization occurs at smaller internuclear separations. Second, at a fixed pulse duration, shorter average time intervals are associated with fragmentation pathways of higher final charge states, as shown in figure 4(b), implying the spatial distribution of the x-ray beam. For example, the transition from (1,1) to (3,1) takes shorter time for pathway of (1,1)–(3,1)–(3,3) -→(7,7) than pathway of (1,1)→(3,1). The former is produced at the centre of the beam transverse spatial distribution corresponding to the most intense part of the beam, while the latter is produced at the wings of the spatial distribution with lower fluence. Although the observed results are intuitive, we show the capability of this methodology to quantitatively demonstrate the time-resolved dynamics and its modification by varying the FEL beam parameters with statistical measurements.
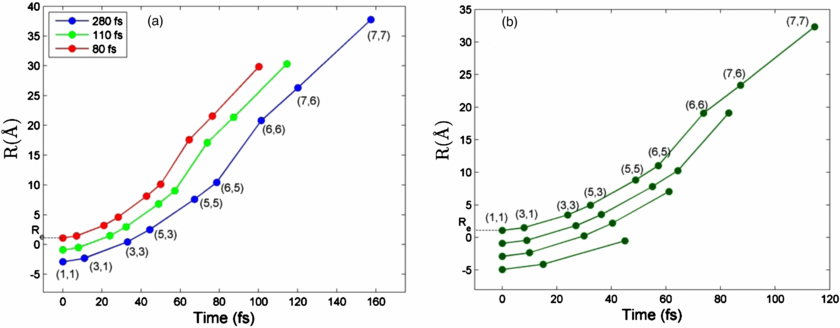
Figure 4. Inernuclear separation and time for each photoionization event for (a) a selected fragmentation pathway at three different pulse durations; (b) different ionization pathways at a selected pulse duration, 110 fs. The dots represent photoionization events. The labels (m, n) correspond to the channel resulting from each PA process.
Download figure:
Standard image High-resolution imageIn summary, we presented an experimental approach to use the multiphoton ionization by x-ray FEL as an intrinsic clock to time-resolve the ionization and fragmentation dynamics in the N2 molecule. We extracted the average KE with measurements of iTOF spectra for different fragment charge states at three different pulse durations. Using a numerical model, we obtained the timing information and the corresponding internuclear separation for each photoionization event. The results show the modification of the dynamics by varying the beam profile with temporal and spatial differences.
3. Probing ultrafast x-ray induced dynamics with correlated and coincident spectroscopy
Tracing the atomic and molecular response to intense x-ray pulses available at x-ray FELs introduces greater experimental challenges as compared to synchrotron measurements or to strong-field studies in the optical and XUV regime. Since absorption of a single x-ray photon typically releases at least two electrons (many more in high-Z elements), multiphoton x-ray processes necessarily involve a large number of electrons in the final state. Therefore, kinematically complete experiments, which provided the most detailed picture of many important light-induced reactions (e.g., single-photon double ionization [44], sequential and non-sequential ionization in strong fields [45, 46], or break-up of diatomic molecules [47]) are currently out of reach at x-ray FELs. Nevertheless, the necessity to understand in detail the behaviour of basic x-ray irradiated systems, such as isolated atoms or simple molecules, within the very limited machine time available for these studies, asks for the most efficient measurement techniques that yield the maximum possible information for a given reaction. For many cases, this can be realized by simultaneous detection of several correlated observables [8]. Within the first three years of LCLS operations, numerous enlightening correlated ion/photon/electron measurements [48–53] were performed using the dedicated multi-parameter CAMP instrument [54]. They stimulated the development of the next generation experimental end-station (LAMP) [7] which is also designed for simultaneous ion, electron and photon detection. In this section, a few illustrative examples are given of correlated ion–photon, ion–electron, and ion–ion measurements which are aimed at revealing the subtleties of ultrafast x-ray induced dynamics in atomic and molecular systems.
3.1. Multiphoton x-ray ionization of xenon: correlated ion and fluorescence spectroscopy
Early LCLS experiments on the photoionization of light elements (e.g., neon, nitrogen) [41, 55, 56] along with extensive theoretical work [57–59] firmly established sequential single-photon absorption as the dominant mechanism of x-ray ionization at intensities up to 1018 W cm−2. Within this picture, the highest charge state produced is typically defined by the last ionic state that can still be ionized with one photon. However, already the first experiments on heavy elements, in this case, xenon, demonstrated a drastic violation of this simple rule [50]. As illustrated in figure 5(a), the highest charge state detected upon the irradiation of xenon atoms by 1500 eV 2.7 mJ LCLS pulses is Xe36 +, although the last ionic state which can be further ionized by a single 1500 eV photon is Xe25 + [60]. Therefore, not only the highest charge state, but even the most abundant charge state observed (Xe30 +) is well beyond the intuitive expectations for sequential ionization model. Here, it should be noted that the data of figure 5, though taken at the same wavelength and maximum pulse energy as the charge states distributions reported in [50], were obtained using a somewhat different experimental configuration. In contrast to the open-geometry ion spectrometer used in [50], a 0.5 mm wide slit placed perpendicular to the LCLS beam propagation was used here to restrict the source volume of the detected ions to the central region of highest fluence within the Rayleigh lengths of the x-ray focus. In addition, x-ray pulses generated from 200 fs electron bunches were used as compared to 80 fs for [50]. Therefore, even though the results presented in figure 5(a) are qualitatively similar to the data published earlier (see figure 1(a) of [50]) and the highest charge state observed is the same, the relative abundances of the low charge states are considerably suppressed as compared to [50].
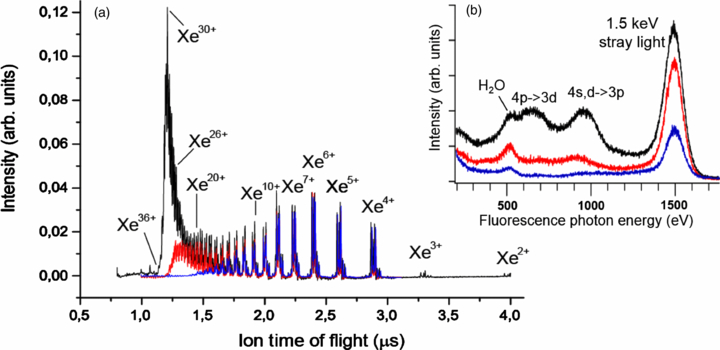
Figure 5. (a) Ion time-of-flight spectra for xenon atoms ionized by 1500 eV LCLS pulses with a nominal pulse length of 200 fs. (b) Simultaneously measured x-ray fluorescence spectra. Black lines: pulse energy (at the LCLS gas monitor detector) 2.7 mJ; red: 1.35 mJ; blue: 0.25 mJ.
Download figure:
Standard image High-resolution imageThe observed unprecedentedly high degree of ionization, well beyond the prediction of simple sequential models [55, 57, 60], implies that another efficient ionization process must play a role here. An insight into the possible mechanism can be obtained from the fluorescence spectra measured simultaneously with the ion charge state distributions (figure 5(b)). At the two lower pulse energies, the spectrum is dominated by stray light and oxygen fluorescence from background water molecules. At the highest pulse energy (black line in figure 5(b)), the characteristic xenon lines appear, correlated with the appearance of higher xenon charge states (compare blue, red and black lines in figure 5(a)). In particular, the broad peak between 800 and 1100 eV photon energy corresponds to the fluorescence energies of 4d→3p and 4s→3p transitions in highly charged xenon ions (above Xe20 +) [50, 60]. However, the energy needed to ionize a 3p electron in these states exceeds 1500 eV. The only mechanism which can efficiently produce 3p vacancies in highly charged ions at this photon energy is the excitation of 3p electrons into densely spaced Rydberg states and unoccupied valence orbitals. Analysis of the wavelength-dependent data, comparison with a theoretical model which includes only bound-free transitions into continuum, and estimated cross sections for possible intermediate resonances convincingly prove that such transient resonant excitations in the multiply charged ions, followed by further Auger decay and other autoionization processes, are responsible for the extremely high charge states observed in figure 5. This resonance-enhanced, or resonance-enabled x-ray multiple ionization pathway has also been observed for krypton atoms [61] and for iodine-containing molecules [62] and is predicted to be a general mechanism for multiple ionization of high-Z elements by intense, short-pulse x-rays.
3.2. Multiphoton x-ray induced fragmentation of molecules: ion–ion coincidence measurements
If a high-Z element is embedded into a molecular system consisting of light atoms, the initial photoabsorption at kilovolt photon energies typically proceeds similar to the case of an isolated atom. However, in a molecule, the Auger cascades, which in this case also trigger dissociation processes, may involve the delocalized valence electrons, thus leading to efficient charge redistribution within the fragmenting system. Thus, coincident measurements of (all) ionic fragments are needed to determine the total final charge state. Comparing ionization of several molecular systems containing a single heavy atom (Se or I) to the ionization of an isolated rare gas atom with similar electronic structure and x-ray absorption cross section (Se and Kr, I and Xe), we found that while the total charge induced on the system is almost the same in both cases, the highest final charge state is considerably higher for an isolated atom [62–64]. As an example, we will discuss here the x-ray photoionization of methylselenol molecules studied by ion–ion coincidence momentum spectroscopy. As shown in figure 6(a), the total charge of the molecule after ionization at 2 keV photon energy closely resembles the charge state distribution of krypton atoms under the assumption that all hydrogen atoms are charged, which is, at least for the highest charge states of Se-C pairs, well justified. However, the highest individual charge state of selenium is Se9 + (compare to Kr16 +), indicating efficient charge redistribution after (multiple) photoabsorption.

Figure 6. (a) Charge state distribution of methylselenol (CH3SeH) molecules and krypton atoms induced by 2keV 5 fs LCLS pulses. The red bars represent the Kr results and the blue bars the sum of Se and C charges measured in coincidence. The highest charge state combination detected is Se9 + + C3 +. The black bars denote the total charge of the molecule calculated assuming that four H+ ions were produced. (b), (c), (d) The KE distributions of Se, C and H ions for three different fragmentation channels. The vertical blue lines indicate the simulated energies for the given charge states assuming instantaneous ionization at the equilibrium geometry of the neutral molecule [63, 64].
Download figure:
Standard image High-resolution imageIn figure 6(b), (c), and (d), the measured kinetic energy distributions (KED) of selenium ions (b), carbon ions (c) and protons (d) detected in coincidence are presented for three different charge state combinations of carbon and selenium. The vertical lines represent the results of a simple simulation of the molecular Coulomb explosion (CE) assuming instantaneous removal of the electrons at the equilibrium bond lengths and angles. In all cases, we experimentally observe a broad energy distribution with its maximum clearly shifting towards higher energies with increasing total charge state. For the lowest charge states, the simulated energies of selenium and carbon ions fit well to the maximum of the KED, whereas they increasingly overestimate the experimental results for the higher charge states. The considerable deviation of the measured KEDs from the simulated CE energies for increasing charge states is a direct measure of the motion of the nuclei on the time scale of multiple ionization. Low charge states are predominantly produced by single-photon absorption, such that fast dissociation starts upon ultrafast core-vacancy decay resulting in kinetic energies of the heavy ions that are in reasonable agreement with the simulations for equilibrium geometry. In contrast, the lightest ionic fragments, protons, are considerably displaced even within this very short time. Therefore, the simulated proton energies are much larger than the measured ones for all charge states (figure 6(d)). These results yield a direct estimate of the radiation damage to the molecule induced by intense femtosecond x-ray pulse.
3.3. Photoelectron spectroscopy of laser-aligned molecules: correlated ion–electron measurements
Because of its high degree of localization, inner shell ionization of molecules provides an efficient tool for imaging molecular structure. The photoelectrons that are created at a specific atom within the molecule can be used to illuminate the molecule 'from within' [65]. They scatter on the neighbouring atoms on their way out and can be uniquely identified in the continuum by their KE. As demonstrated e.g. for molecules adsorbed on surfaces, the measured diffraction pattern of these photoelectrons then yields structural information [66, 67]. Combined with the short duration of x-ray FEL pulses, the photoelectron diffraction technique has the potential for taking snapshots of the time-evolution of photo-induced chemical reactions. However, in order to be able to extract this information from the recorded photoelectron angular distributions, the reference frame of the molecule has to be known. The molecular axes can be determined from the measurement a posteriori, for example in an angle-resolved photoelectron-photoion coincidence experiment [65, 68]. For the case of methylselenol molecule discussed above, it was shown that orientation of the molecule can be defined from the angles of the detected ionic fragments [64]. However, since this technique is applicable only for rather small molecules and requires less than one ionization event per pulse, an alternative approach based on active laser alignment of molecular ensembles has been attracting increasing attention, especially in view of rather low repetition rates of the currently operating x-ray FELs (<120 Hz). Nevertheless, even in the latter case, the diagnostic of molecular alignment is based on measuring the angular distribution of the ionic fragments [69], typically performed employing the so-called velocity map imaging (VMI) [70]. Implementation of this alignment and diagnostic technique using a multi-parameter instrument such as the CAMP [54] or LAMP [7] experimental end stations at LCLS thus enables simultaneous photoelectron measurements and diagnostic of molecular alignment and thereby allows for correlated ion–electron measurements.
Figure 7 illustrates the concept of such an experiment, where 1-ethynyl-4-fluorobenzene (pFAB) molecules were adiabatically aligned using a nanosecond Nd:YAG laser and probed by photoelectron diffraction at different photon energies above the fluorine K-edge [53]. Both, photoions and photoelectrons are measured simultaneously using a double-sided VMI spectrometer [54]. Note that in contrast to the results presented in the previous sub-section, this experiment is performed on an ensemble of molecules and, thus, does not yield real coincident data. Molecular alignment is probed by measuring the angular distributions of F+ ions. Without the alignment laser (figure 7(b)), the distribution is almost isotropic, whereas in the presence of the Nd:YAG beam (figure 7(a)), the ion emission pattern is strongly aligned along the laser polarization direction yielding an alignment parameter of <cos 2θ > 2D = 0.85. Figure 7(c) displays the (inverted) photoelectron VMI image from an aligned ensemble of pFAB molecules. The fluorine 1s photoline of interest is visible as a narrow dipole-like structure in the upper and lower part of the image centred at 51 eV, whereas the central region is dominated by low-energy electrons, mostly resulting from shake-off type processes and Auger cascades. This low-energy contribution is further enhanced by the Nd:YAG ionization of highly-excited molecular states produced by the x-ray pulse [71]. Since the nanosecond Nd:YAG laser pulse used for adiabatic alignment is centred on the femtosecond x-ray pulse, the Nd:YAG field is present for a few ns after the x-ray pulse and can thus post-ionize molecules that have been excited by the x-rays. Subtracting the VMI photoelectron images obtained from aligned and isotropic ensembles and measuring these difference maps for different photon energies (and, thus, for different de Broglie wavelengths of the outgoing photoelectron waves), one can relate the measured photoelectron angular distribution differences between aligned and unaligned molecules to the molecular geometry by comparing the outcome with theoretical simulations [53]. As shown in [53], this technique opens the way towards direct time-resolved imaging of molecular transition states in UV pump–x-ray-FEL probe experiments.

Figure 7. (a) VMI image of F+ ions produced by the ionization of an adiabatically aligned ensemble of pFAB molecules by 743 eV LCLS photons. (b) Same for the isotropic ensemble. (c) The inverted VMI photoelectron image measured simultaneously with the ion data of figure 7(a). Polarization of both, LCLS and Nd:YAG beam is parallel to the y-direction. The radial axis shows the electron KE.
Download figure:
Standard image High-resolution image4. Probing molecular transformation via time-resolved x-ray dissociative ionization: 1,3-cyclohexadiene isomerization
The dominant relaxation pathway after core ionization of lighter elements, including carbon, proceeds through the emission of Auger electrons. Molecules composed of lighter elements are thus left multiply charged upon ionization and subsequent Auger decay. Multiply ionized molecules typically undergo Coulomb repulsion-mediated dissociation that results in several positively charged fragments. The distribution of the ion fragments, their kinetic energies and momenta carry information about the instantaneous geometry of the parent molecule at the onset of ionization [72].
With the recent advent of short-pulse x-ray sources, which can be synchronized with optical lasers, it became possible to use dissociative ionization initiated by core absorption as a probe in time-resolved experiments [24, 25, 73, 74]. Time-resolved dissociative ionization can probe transient species in molecules that are undergoing photoinitiated geometry changes. Often it is neither necessary nor practical to completely atomize the molecule in order to reconstruct the full geometry. Rather, partial fragmentation can be informative when a change in the fragmentation patterns reflects a certain aspect of the change in the molecular geometry.
1,3-cyclohexadiene (CHD) is a molecule convenient for testing new experimental techniques for probing structural changes [75]. Absorption of UV radiation initiates the ring opening in this molecule to form 1,3,5-hexatriene (HT). Figure 8 shows the states involved in this process. CHD ring opening is a prototypical example of conrotatory photoinitiated ring opening often used to illustrate the Woodward–Hoffman rules [76]. The same motif appears in important biochemical processes such as vitamin D synthesis. The CHD/HT isomerization has received significant attention in experiments and calculations that seek to investigate the role that the non-Born–Oppenheimer interactions play in the photorelaxation [24, 75, 77–96]. It is now known that the ring opening is complete in less than 200 fs, as a result of nonradiative relaxation through a conical intersection between the S1 and S0 states [75, 81, 84].
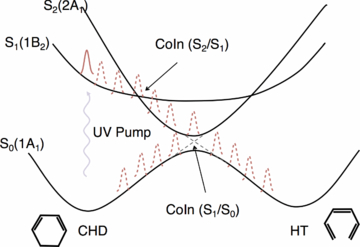
Figure 8. Schematic representation of the states involved in the photoinitiated ring opening of 1,3-cyclohexadiene (CHD) to form 1,3,5-hexatriene (HT). A UV pulse launches a wavepacket on the spectroscopically bright 1B2 state. The wavepacket rapidly crosses onto the 2A1 potential energy surface where it is accelerated towards the S1/S0 conical intersection. The ring opening occurs in under 200 fs.
Download figure:
Standard image High-resolution imageIn the present study, we report new data from an experiment performed in 2010 at the LCLS at the SLAC National Accelerator Laboratory by the AMO14110 collaboration [97]. The experiment explored several aspects of dissociative ionization initiated by x-ray photoabsorption. Much of the experimental time was devoted to studies of isomerization, where the x-ray-induced fragmentation was the probe for CHD ring opening initiated by absorption of a UV photon. Briefly, an ultrafast UV laser pulse (50 μJ, 266 nm, ∼50 fs) initiated the ring opening in CHD. A collinear time-delayed x-ray probe fragmented the molecule. Positive ion fragments were detected by either VMI or iTOF. Processing of the VMI images yields the KE released in the fragmentation, and the iTOF data show the ion charge and mass distribution. Further details on the experimental setup have been described in the initial publications, which analysed the fragmentation induced by transient x-ray absorption alone following the UV pump [24]. The present report extends this work by analysing x-ray-induced fragmentation when a strong infrared laser field is also present during the x-ray absorption. We find that the laser field changes the fragment distribution, and discuss the significance of these changes.
Transient fragmentation of the CHD ring opening reaction using only a strong infrared laser field has been studied extensively. The parent CHD and daughter HT have markedly different laser-induced fragmentation patterns. Differences also exist when the probe is an ultrafast x-ray pulse, but the changes are less pronounced [24, 81]. Both x-rays and infrared laser probes show an increase in the H+ ejection for HT [24]. In addition, for the x-ray probe the average KE of the fragments is larger for the HT geometry (see figure 9) [24]. This change is observed on a ps timescale.
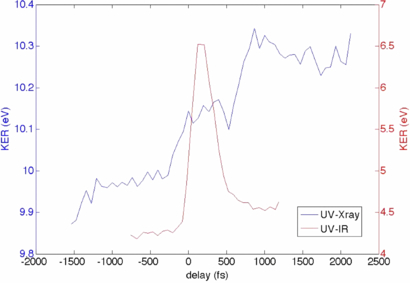
Figure 9. Evolution of the KE averaged over all ion fragments with pump–probe time delay for x-ray probe (blue; axis on the left) and IR probe (red; axis on the right). Pump wavelength was 266 nm in both cases. See [24] for more details.
Download figure:
Standard image High-resolution imageTime-resolved pump–probe studies at FELs have been limited by poor synchronization between the optical lasers and the x-ray pulses [25]. This low temporal resolution prevents a detailed view of transient dynamics of ring opening, but some information about the transient state can still be obtained. Figure 9 overlays fragment KE data with an x-ray probe and an infrared probe. In both cases the average KE increases when the ring opens; however, the strong infrared laser has a sharp transient maximum in the average KE, which is not observed for the x-ray probe [24].
This comparison suggested that the strong transient enhancement is due to a process inherent to the strong-field fragmentation. More detailed analysis of the fragment distribution patterns in the strong-field case reveals that the delayed transient increase in the energetic fragments is correlated with formation of the dication CHD2 + [83, 85]. This delayed enhancement in the cross section for double ionization occurs during the passage of the molecular geometry through the vicinity of a known conical intersection between the ground and first excited states in the molecule [83, 85].
The x-ray probe also leads to formation of the molecular dication prior to fragmentation, but the mechanism is core-vacancy formation followed by Auger electron emission, not strong-field ionization. There is no apparent transient enhancement in the x-ray-initiated process as the molecule passes through a conical intersection [24]. This is because the x-ray absorption cross section is not affected very much by the molecular geometry, and the strong x-ray fields, unlike strong infrared fields, do not perturb the valence electrons. The x-ray probe fragmentation pattern therefore provides distinctly different information than the infrared laser probe.
To investigate further the different mechanisms of IR and x-ray-induced photofragmentation, we performed an experiment in which the UV pump was replaced by a strong IR laser, to see how an intense external IR field modifies the ion-fragment distribution from ground-state CHD. The experiment was performed under the same condition as the UV-x-ray experiment discussed above, except that in this case an 800 nm pulse, ∼50 fs long, collinearly propagating with the x-ray beam was used instead of the UV beam. The delay between the x-ray and the IR pulses was scanned over a ∼2 ps range and the ion fragments were collected using the iTOF detector.
A subset of fragments displays an increase in production that peaks when the timing of the two pulses roughly coincides. This is the case for example for H+, H , C+, CH+, C2 +, etc, while some of the CnH
, C+, CH+, C2 +, etc, while some of the CnH fragments, particularly those with multiple hydrogen atoms, such as CH
fragments, particularly those with multiple hydrogen atoms, such as CH , CH
, CH , CH
, CH , C2H
, C2H , etc, display depletion when the IR pulse arrives after the x-ray pulse. Figure 10 compares the time-dependence of the production of H+ and CH
, etc, display depletion when the IR pulse arrives after the x-ray pulse. Figure 10 compares the time-dependence of the production of H+ and CH ions. Temporal overlap between the infrared and x-ray beams was generally maintained using the method described in [25], but in figure 10, we were unable to employ this method. Instead we have set T = 0 at the point in the plot where the H+ production has a maximum and the parent peak displays a minimum.
ions. Temporal overlap between the infrared and x-ray beams was generally maintained using the method described in [25], but in figure 10, we were unable to employ this method. Instead we have set T = 0 at the point in the plot where the H+ production has a maximum and the parent peak displays a minimum.
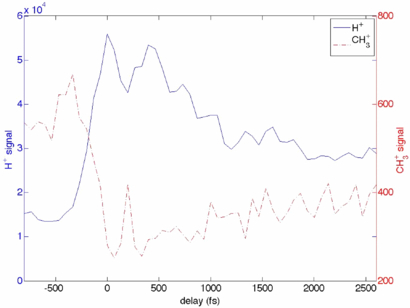
Figure 10. Intense IR field alters the distribution of the x-ray produced ion fragments in a time-delay dependent manner: shown is the dependence of the H+ (blue, full line) and CH (red, dashed line) signals on the time delay between the x-ray and the IR pulses. Ions were detected using a in iTOF detector.
(red, dashed line) signals on the time delay between the x-ray and the IR pulses. Ions were detected using a in iTOF detector.
Download figure:
Standard image High-resolution imageEven for molecules smaller than CHD it is challenging to model ab initio their relaxation upon core ionization when they are undergoing a coupled change in the nuclear geometry and electronic state symmetry. A calculation of the distribution of the dicationic states formed by the Auger relaxation shows that this distribution is sensitive to both the electronic excitation and the nuclear geometry change [24]. This calculation showed that a significant number of states, differing in the excess energy available for the fragmentation, are populated by the Auger process. The distribution of the states differed depending whether the Auger process occurred in the ground state CHD, its first excited state, or in HT [24]. Understanding the nature of the distribution of the dicationic states that are populated by the Auger relaxation is important for interpreting the time-resolved x-ray dissociative ionization of evolving molecular structures. This study can help to validate models of this process.
We propose that several processes contribute to the signal observed in figure 10. When both the strong field and the x-ray pulse arrive at the same time, the Auger decay occurs between the field-dressed states. The field dressing modulates the transition intensities for different Auger pathways, thus affecting the distribution of the dicationic states from which the ion fragments originate. Further, when the IR pulse arrives after the x-ray pulse, it can induce transitions between different short-lived dicationic states, modifying their distribution [98]. Finally, strong field can lead to further fragmentation of the primary fragments formed in the relaxation upon the core ionization. Our hypothesis that the secondary IR fragmentation plays a role is supported by the observation that the depletion occurs more likely in many-particle fragments while a corresponding increase appears in fewer-particle fragments. The temporal resolution in this case does not permit separation of the different processes discussed above. However, it is unambiguous that the strong field modifies the fragment distribution in a manner that is sensitive to the delay between the x-ray and IR pulses. Further studies with ion–ion coincident detection would provide more details about the mechanisms of these competing processes.
The early LCLS experiments performed in 2010 that are discussed here indicate that the x-ray initiated dissociative ionization reflects a change in the molecular geometry [24]. However, due to the large number of dicationic states populated by the Auger relaxation, the relationship between the fragmentation patterns and the molecular geometry is more difficult to interpret than in the case of strong-field dissociative ionization. The two probing regimes are complementary. Both will benefit from ion–ion coincident detection that will permit separation of competing processes. Currently the biggest obstacle to more widespread use of the coincident detection schemes in FEL experiments is the relatively low repetition rate of the FEL x-ray sources. This low repetition rate necessitates longer data acquisition, which is difficult to execute in limited time allocated for the FEL experiments. Low temporal resolution, which hampered early FEL experiments described here, has since been mitigated at LCLS by implementation of techniques that measure the arrival of the x-ray pulses with respect to the laser pulse on each shot [99, 100]. This permits the data to be re-sorted in the post processing with improved temporal resolution. Time-resolved probing of evolving molecular structures will benefit greatly from an increase in the repetition rate of the FEL x-ray sources, improvements in the synchronization between the x-ray pulses and the laser pulses, and greater abundance of the FEL x-ray sources.
5. Probing ultrafast dynamics in extended and nanoscale systems
FEL can deliver extremely intense, coherent x-ray flashes with femtosecond pulse length, opening the door for imaging single nanoscale objects with atomic resolution in a single shot. All matter irradiated by the intense x-ray pulses at peak intensity, however, will be transformed into a highly-excited non-equilibrium plasma within femtoseconds. During the x-ray pulse complex electron dynamics and the onset of structural disorder will be induced, leading to a constantly changing sample on the femtosecond time scale. Investigating the electron and ion dynamics in nanoplasma poses specific challenges due to the hundreds to thousands reaction products and new concepts are needed.
In a time dependent study about the x-ray absorption dynamics in small argon clusters a combination of precisely tuned x-ray pulse length [102] and ion spectroscopy was used and the concept of x-ray induced transparency increase XITI was introduced [101]. The iTOF spectra in figure 11 of clusters irradiated with femtosecond x-ray pulses demonstrate that the intensity of high charge states increased compared to the lower charge states for a longer pulse length with the same pulse energies, similar to earlier observations about x-ray induced transparency in atoms [55] and molecules [41]. A quantitative analysis of the x-ray induced transparency in clusters is, however, not as straight forward possible due to rapid recombination of the nanoplasma prior to the reaction product detection in the spectrometer [103]. For a quantitative analysis of the effect in clusters, the XITI has to be used which is a measure of the time dependent component in the absorption. The XITI concept is based on the fact that the secondary (recombination) processes are slow compared with the femtosecond x-ray pulse and thus, the same number of absorbed photons will lead to the same final ion yield spectrum and average charge state. Measuring the two dimensional parameter space of pulse length and pulse intensity allows one to compare how many more photons are needed for a shorter pulse to produce the same average charge state of a longer pulse and thus, how much more transparent the cluster for a short versus a long x-ray pulse is. Specifically, for the 〈N〉 = 1600 cluster discussed in figure 12 60% more pulse energy is needed to produce the same average charge state with a 30 fs pulse compared with the result for a 85 fs pulse. In other words, an argon cluster of 1600 atoms is 60% more transparent for an intense 30 fs x-ray pulse compared to an 85 fs one, meaning that the XITI in this case is 60%. By that means it was found that there exists a clear cluster size dependency for the XITI and thus, that larger clusters absorb x-rays less efficiently compared to smaller ones and atoms [101]. The reason for that is that the inner shell ionization dynamics are strongly influenced by the atomic environment of the cluster. The explanation for the decreased x-ray ionization efficiency in extended samples compared to individual atoms is that the Auger lifetimes are increased in the massively ionized nanoplasma, leading to increase core-level bleaching [101, 104].
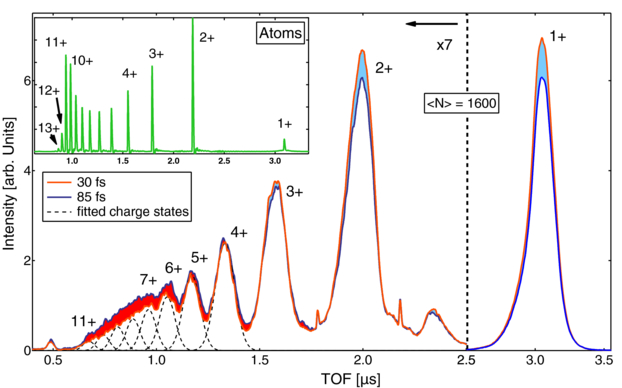
Figure 11. TOF spectra from argon clusters for two different pulse lengths. The difference between long and short pulses is indicated in orange (higher intensity for long pulses) and blue (higher intensity for short pulses). Inset: atomic argon TOF spectrum for the same pulse energy and 50 fs pulse length. Compared to the atomic spectrum the ion peaks from the cluster are shifted and broadened due to the KE release from the cluster explosion. Reprinted with permission from [101]. Copyright 2012 by the American Physical Society.
Download figure:
Standard image High-resolution image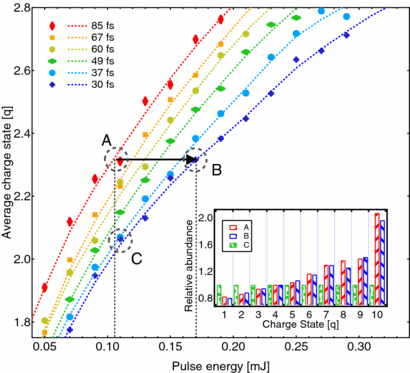
Figure 12. Average charge state of the detected ionic cluster fragments versus x-ray pulse energy for clusters with an average size of 〈N〉 = 1600 atoms. The different colours indicate different pulse length. The diamond shaped markers are binned data points and the dashed lines interpolated curves. The black horizontal arrow marks the XITI from a 30 to a 85 fs pulse. Inset: Abundances of charge states at point A, B and C. For better visibility single charge states are normalized to the value of point C. Reprinted with permission from [101]. Copyright 2012 by the American Physical Society.
Download figure:
Standard image High-resolution imageAnother opportunity for investigating the ultrafast dynamics in x-ray excited clusters and nanoplasma is the analysis of their scattering patterns. The intense x-ray pulses from a FEL, delivering typical intensities of 1012 photons in a hundred femtosecond pulse allow taking single particle images with single shots. Coincident scattering and spectroscopy can yield unique insight into the x-ray induced nanoplasma dynamics [48, 105]. Resonant scattering of large xenon clusters at the giant Xe 4d resonance has revealed strongly power-density dependent scattering patterns, yielding insight into the complex interplay between excitation and scattering in nanoscale objects with x-ray pulses [105]. The scattering signal is affected by the changes in the electronic structure of the sample, leading to modifications of the scattered light even though the particle geometry remains the same on the time scale of the interaction with the laser pulse. Conversely, the information carried by the scattering signal can be exploited to gain insight into transient electronic states on the femtosecond time scale. For simple systems such as clusters the scattering data can be modelled with Mie theory. The variable parameters for the Mie fits are the particle size and the wavelength-dependent refractive index n. In the x-ray spectral regime the refractive index is commonly expressed as n = 1 − δ + iβ. The real part 1 − δ is a measure of the phase shift of the scattered wave and connected to how efficient the whole atom scatters compared to the Thomson scattering of a single free electron. The imaginary part β describes the absorption of the system and is directly proportional to the photo absorption cross section σ. In consequence, by measuring the scattering profiles and modelling with Mie theory, optical constants and thus information about the electronic structure of a particle can be determined. In figure 13 the absorption parameters deduced from scattering patterns taken with increasing power density are shown and compared to TOF data recorded under similar conditions. The data show that the absorption deduced from the scattering patterns is significantly higher compared to the spectroscopy data which can be correlated to resonant ionization processes in the particle during the x-ray pulse [105]. The scattering pattern is only generated during the pulse and therefore it is susceptible to the x-ray pulse-induced transient changes in the particle electronic structure. Vice versa, the scattering signal carries information about electronic structure changes during the light–matter interaction. Thus, single-shot single particle scattering yields insight into ultrafast processes in highly-excited systems where conventional spectroscopy techniques such as TOF mass spectroscopy are inherently blind as the ions and electrons are detected long after reaction and as they are subject to secondary processes such as plasma recombination. Therefore, scattering experiments with intense ultrashort x-ray pulses open new opportunities for investigation of transient electronic configurations of highly-excited states of matter, non-equilibrium plasmas, and dense exotic systems, in addition to being key for solving structural information problems.

Figure 13. Particle absorption parameter β deduced from the scattering patterns. Right: modelled relative absorption contribution of the dominant ions based on atom and cluster target TOF spectra recorded with similar laser conditions. Reprinted with permission from [105]. Copyright 2012 by the American Physical Society.
Download figure:
Standard image High-resolution image6. Soft x-ray split and delay
Several methods are available to provide two ultrafast pulses for pump–probe experiments using soft x-rays from the LCLS. The first method used an external optical laser to pump or excite the target with the x-ray pulse arriving at some later time to probe the temporal evolution of the system on the ultrafast time scale. Time-resolved electron spectra from aligned N2 molecules were first obtained by this method at LCLS [74] in 2009 and it has subsequently been used in numerous other studies on molecular dynamics and other fields. While the method is powerful and facilitates the use of ultrafast x-ray pulses from the LCLS to study dynamic systems, it is limited by the uncertainty in the arrival times of the two pulses. Due to the extended length of the LCLS FEL along with jitter in the electron acceleration, the resulting uncertainty of the arrival time of the x-ray FEL pulse relative to the optical laser pulse is at best around 280 fs FWHM [25]. To overcome this large band of arrival time uncertainty an optical/x-ray cross correlator has been developed and deployed in further experiments to measure the relative arrival times of the two pulses to within 50 fs FWHM [27]. This so-called 'time-tool' only measures the relative arrival times of the optical and x-ray pulses allowing data to be sorted on a shot-by-shot basis into smaller uncertainty bins but does not control the time difference, giving rise to a large range of time delays being stochastically sampled by the inherent jitter in the pulse arrival times.
A novel accelerator based method to generate two time-separated x-ray pulses has also been deployed at the LCLS utilizing a slotted foil [106]. The foil, with two slots in it with varying separation forming a V shape, is inserted into the electron beam in the centre of the second bunch compressor chicane in the LCLS accelerator. The bunch compressor is a magnetic chicane that is used to shorten the bunch in its longitudinal direction, corresponding to time. In the centre of the chicane, after the electrons have passed through two of the four magnets, the electron bunch has been turned on its side, with the head and tail in the transverse direction relative to its motion and time is encoded in the transverse direction. Electrons passing through the foil have their emittance spoiled by scattering through the material while those passing through the slots in the foil are unaffected and maintain their original emittance. When the electron beam subsequently passes through the undulator only the portions of the electron bunch with the original emittance are able to lase and produce intense FEL radiation. Electrons that passed through the two slots in the foil therefore produce two FEL pulses separated in time proportionally to the distance between the slots in the slotted foil in the bunch compressor. Time separation of the pulses is controlled by the position where the electron beam passes through the angled slots. This method has been used to produce two time separated x-ray pulses with stable time separation for experiments at the LCLS. The principle disadvantage of this method for producing two ultrafast x-ray pulses arise from the independent production of FEL radiation from the two portions of the electron bunch that passed through the slots in the foil. Since the two portions of the electron bunch lase independently they are not coherently related and while the total intensity can be determined it is not possible to measure the relative intensities of the two x-ray pulses.
To overcome the shortcomings of these methods we have developed an optical x-ray split and delay (XRSD) system to split the incoming x-ray pulse into two time separated pulses. Split and delay units are commonly used in optical laser systems where a laser pulse can be separated by a partially transmissive mirror into two paths, the time delay controlled by varying the length of one of the paths and the paths of the two pulse merged and sent on to the sample for a pump–probe experiment. This method has recently been extended into the vacuum ultraviolet (VUV) wavelength range using multiple reflective mirrors at the FLASH VUV/soft x-ray FEL [107]. These devices are limited by the large angles at which the mirrors reflect the beam to wavelengths greater than 6 nm. Using a split normal incidence multilayer focusing mirror, two 38 eV time separated x-ray pulses were produced to study molecular dynamics at the FLASH FEL [108]. Multilayer mirrors work only at specific wavelengths, however, and do not provide a general purpose device. Below we describe a device that provides two x-ray pulses separated by up to 200 fs with sub-fs time resolution and functions over the full soft x-ray energy range of LCLS, 250 1800 eV. The upper limit of photon energy is due to the cut-off of reflectivity at ∼1800 eV of the silicon KB mirrors. The device will be described in greater technical detail in a forthcoming publication [109].
In order to function over the full soft x-ray range of the LCLS, the mirrors in our soft XRSD unit are operated at angles of less than 13.5 mrad relative to the incoming beam, much shallower angles of incidence than in the devices described above for the VUV range. Owing to this shallow incidence angle and the divergence of the x-ray FEL beam to a FWHM of ∼1–2 mm at the wavelengths of interest, a versatile eight mirror device with sufficient clear aperture would require over 5 m of length along the beamline, space that is not currently available. Instead we chose to develop a two mirror compact device that requires only ∼1 m of space along the beamline. The principle of the devices operation is illustrated schematically in figure 14. Briefly, two mirrors are situated along the beam path such that the first mirror intercepts a portion of the beam and deflects it towards the interaction region. The second mirror intercepts the remaining portion of the beam somewhat downstream of the first mirror and deflects that portion through a slightly larger angle to intersect the beam from the first mirror in the interaction region of the experiment. In order to split the beam without significant losses, the first mirror was polished to its trailing edge where a short chamfer allows the beam not reflected by the mirror to pass over the edge to the second mirror. Rather than translate the second mirror along the beam path to intersect the second (delayed) portion of the incident beam this design incorporates an extended second mirror requiring only vertical motion and pitch to properly intercept the beam and reflect it towards the interaction region. Many x-ray pump–x-ray probe experiments require high intensity to probe nonlinear phenomena requiring the use of focusing optics to achieve small spot sizes and therefore high intensity in the interaction region. This XRSD unit is located between the KB focusing optics in the LCLS AMO beamline and the experimental end-station. While this extends the focal length of the KB optics from 1.1 to 2.3 m, and hence increases the minimum achievable spot size in the interaction region by about two times, it simplifies the optical scheme and changes required to insert and remove the XRSD. This arrangement places stringent alignment and stability tolerances on the two mirrors in the XRSD to achieve and maintain overlap between two spots of approximately 2 μm in diameter in the interaction region. This was achieved with a mechanically simple yet precise system that uses stepper motor driven linear stages, piezo actuators for fine control and encoder and interferometer read-back of the positions of the mirrors along with their angles. To verify that the mechanical systems were meeting the required precision several procedures were used during commissioning of the XRSD to verify that the x-ray beams from the two mirrors were overlapped in the interaction region of the experimental end-station. First an optical microscope with ∼3 μm resolution was used to view a YAG crystal in the interaction region of the spectrometer. Motion stages controlling the vertical position and angle of the two mirrors were calibrated to yield overlapping spots on the YAG screen over a range from 0–200 fs delay. Following this calibration a series of imprints of the x-ray pulse in PbWO4 crystals were measured to verify the overlap with greater precision [110]. Finally, ion spectra of argon were measured at different pulse delays. Yields of higher charge states increased when the two pulses overlapped spatially in the ion spectrometer due to the higher fluence on each atom from the two pulses rather than each pulse hitting a different sample of atoms in the spectrometer. In this means the motion of the mirrors was calibrated and verified to yield spatially overlapping pulses of x-rays.

Figure 14. Schematic of the two mirror soft XRSD unit. The first mirror, polished up to its trailing edge, is inserted into the converging x-ray beam to deflect half of it towards the interaction region. The second mirror intercepts the remaining beam at a distance downstream that is approximately equal to the delay between the two pulses and deflects it at a larger angle to overlap with the first beam in the interaction region.
Download figure:
Standard image High-resolution imageAn optical laser-x-ray pulse cross correlator was used to verify the temporal separation of the two pulses from the XRSD mirrors [27]. A thin silicon nitride, Si3N4, film was placed downstream of the interaction region and oriented to be 45° to the incident x-ray beams about the vertical axis. Mirrors in the XRSD were set to vertically separate the beams with the beam from the mirror first above the beam from the second. Laser pulses from a Ti:sapphire laser were passed through normal to the film coincident in time with the x-ray pulses and the transmission through the Si3N4 film recorded on a CCD camera for each pulse. The resulting false colour intensity map is shown in figure 15 where two green–red features are observed. These features correspond to the two x-ray pulses from the XRSD hitting the Si3N4 membrane, modifying its electronic and optical properties and thus changing the absorptivity of the film for the 800 nm light. The first pulse, arriving 150 fs earlier than the second pulse at the Si3N4 film has progressed further along the angled foil than the second pulse when the optical laser pulse arrives and thus is encoded in the image at a different horizontal location than the second pulse. A separate calibration of time to distance relationship was performed by varying the delay between the laser and FEL pulses.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 15. Image of the Ti:sapphire laser light transmitted through a thin Si3N4 membrane when illuminated with two x-ray pulses separated by ∼150 fs from the soft XRSD unit. Transmission of the 800 nm light through the film increases in areas illuminated by x-rays, spatially encoding the arrival time of the x-rays relative to the laser pulse in the horizontal direction.
Download figure:
Standard image High-resolution image{kind=link}
In summary, a two mirror XRSD device for use with the soft x-ray beamlines at the LCLS was described. The device splits the incoming FEL pulse with an edge polished mirror, delays the remaining portion of the pulse and recombines them in the interaction region of the experimental end-station. The performance of the instrument was demonstrated from calibration data measured during its commissioning.
7. Future research possibilities with FELs
Further technological developments on FELs and new techniques have been carried out to extend the scientific capabilities enabled by FELs. The FERMI@Elettra light source (Italy) which is a laser-seeded setup is operational since last year and enables new types of experiments due to the stability and spectral resolution of the facility. LCLSII which is under construction will also deliver self-seeded FELs with a spectral bandwidth of ∼0.5 eV [7]. With the seeding technique, the photon energy stability and longitudinal pulse coherence will be greatly improved, opening up experimental approach with high photon-energy resolution as well as high pulse energy and temporal stabilities. Future FELs are promising to provide photon beams with improved capability, such as simultaneous multi-beamline settings, higher repetition rates which will make possible fast data collection in coincident experiments and x-ray diffraction experiments, higher beam intensities which can lead to efficient non-sequential photoaborption at core levels, and shorter pulse duration that enables investigation on a-few-femtosecond electronic dynamics. The higher flux of light sources has led to efficient depletion of core-hole electrons, bringing the realization of double-core-hole spectroscopy. Other capabilities have been or will be added, such as the capability of changing x-ray polarization, enabling experiments with non-linearly polarized x-ray beams. There are several FEL facilities that have been built or under construction, such as SACLA (in operation, Japan), Shanghai FEL (China), Swiss FEL (Switzerland), European XFEL (Hamburg, Germany), Pohang (PAL) FEL (South Korea). Furthermore, advanced end-stations at beamlines have been or will be built to combine the power of different detection tools and enable investigations of correlations between structural dynamics and various interaction mechanisms. For example, advanced instrumentation, such as CAMP (FLASH, Germany) [54] and LAMP (LCLS, USA) which was recently commissioned, is capable of carrying out measurements of ion, electron, and photon signals simultaneously, even in a coincident manner, and therefore combine the strength of ion/electron spectroscopy, fluorescence spectroscopy and diffraction imaging [7].
Acknowledgments
This work was funded by the Department of Energy office of Science, Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences under grants no. DE-FG02-92ER14299.A002 and no. DE-FG02-86ER13491. Part of this work was funded by the PULSE Institute at Stanford University and the SLAC National Accelerator laboratory. VP and PB were supported by a grant from the National Science Foundation. DR acknowledges support from the Helmholtz Gemeinschaft through the Young Investigator Program. We acknowledge the Max Planck Society for funding the development and operation of the CAMP instrument within the ASG at CFEL. Part of this research was carried out at the LCLS at the SLAC National Accelerator Laboratory. LCLS is an Office of Science User Facility operated for the US Department of Energy Office of Science by Stanford University.