


ABSTRACT
We report oscillator strengths for six strong vibrational bands between 105.0 and 115.2 nm, associated with transitions from the v = 0 level of the X1Σ+ ground state to the v = 0 and 1 levels of the B1Σ+, C1Σ+, and E1Π states, in 12C16O, 12C17O, 12C18O, 13C16O, and 13C18O. These measurements extend the development of a comprehensive database of line positions, oscillator strengths, and linewidths of photodissociating transitions for all astrophysically relevant CO isotopologues. The E–X bands, in particular, play central roles in CO photodissociation and fractionation models of interstellar clouds and circumstellar disks including the early solar nebula. The resolving powers of the room-temperature measurements, R = 300,000–400,000, allow for the analysis of individual line strengths within bands; the measurements reveal J-dependences in the branch intensities of the C(v = 0,1)–X(0) and E(v = 0,1)–X(0) bands in all isotopologues. Minimal or no isotopologue dependence was found in the f-values of the C(v = 0,1)–X(0) and E(v = 0,1)–X(0) bands at a ∼5% uncertainty level. Revised dissociation branching ratios for the C(v = 0,1) and E(v = 0,1) levels are computed based on these f-values. The weak isotopologue dependence of the f-values presented here eliminates this mechanism as an explanation for the large 17O enrichments seen in recent laboratory photolysis experiments on CO at wavelengths from 105 to 108 nm.
Export citation and abstract BibTeX RIS
1. INTRODUCTION
The photochemistry of carbon monoxide affects the structure and evolution of many astronomical environments, including interstellar clouds, circumstellar disks around newly formed stars, and the envelopes surrounding highly evolved stars. When in the presence of a strong ultraviolet field, the primary destruction mechanism for interstellar and circumstellar CO and its isotopologues is photodissociation, which is entirely governed by discrete line absorption into predissociating levels in the wavelength range 91.2–111.8 nm. Because the CO spectrum consists primarily of resolved line features, self-shielding effects in high-column density environments (e.g., diffuse clouds: Federman et al. 2003; Sheffer et al. 2007; circumstellar disks: Smith et al. 2009) can lead to strong isotopic fractionation signatures in both CO and elemental oxygen and carbon (e.g., Bally & Langer 1982; Sheffer et al. 2002; Sonnentrucker et al. 2007).
CO self-shielding in the solar nebula has been invoked (Clayton 2002; Yurimoto & Kuramoto 2004; Lyons & Young 2005) to explain the unusual oxygen isotope ratios observed in the earliest solar system condensates, viz. calcium–aluminum inclusions (CAIs) in primitive meteorites. Analysis of solar wind collected by the NASA Genesis mission (McKeegan et al. 2011) showed that the Sun has an oxygen isotope anomaly similar to that of the isotopically lightest CAIs, a result that is consistent with CO self-shielding in the early solar nebula or parent cloud. Astronomical observations of CO isotopologue ratios (Sheffer et al. 2002; Smith et al. 2009) although valuable, are of insufficient precision to quantitatively address the hypothesis that CO self-shielding is responsible for the oxygen isotope ratios seen in CAIs in primitive meteorites. A comprehensive database of line positions, oscillator strengths (f-values), and linewidths for all relevant CO isotopologues is needed to assess this hypothesis and for the development of models of astrophysical environments. For example, the solar system meteoritic oxygen isotope measurements are extremely precise, but the existing CO photoabsorption data are inadequate for accurate photodissociation calculations needed to quantitatively evaluate the nebular CO self-shielding theory. Despite considerable experimental and theoretical efforts, significant uncertainties and gaps remain in the CO isotopologue spectroscopic database.
In 1988, van Dishoeck and Black developed a detailed CO photodissociation model that includes depth-dependent and isotope-selective photodissociation rates. Visser et al. (2009) updated and extended that widely used model, incorporating results from the subsequent 20 years of laboratory measurements. The detailed surveys of high-resolution line positions (12C16O, 13C16O, 12C18O, 13C18O) and medium- and low-resolution band f-values (12C16O, 13C16O) between 91 and 101 nm by Eidelsberg & Rostas (1990) and Eidelsberg et al. (1991) served as one primary database, supplemented by measured f-values for selected bands from the work of Federman et al. (2001) and Eidelsberg et al. (2004b, 2006). Visser et al. (2009) also incorporated the results of a suite of laser-based measurements by the Ubachs group at Vrije Universiteit Amsterdam (Ubachs et al. 1994, 2000; Cacciani et al. 1995, 1998, 2001, 2002; Cacciani & Ubachs 2004; Eikema et al. 1994). These measurements established linewidths, line positions, and term values for low-J rotational lines in the relatively sharp transitions to low-lying Rydberg states and in selected bands at higher energies; the work included measurements of the isotopologues 12C16O, 12C17O, 12C18O, 13C16O, and 13C18O.
Notwithstanding the progress represented by the spectroscopic compilation of Visser et al. (2009), the current laboratory database is insufficient for a comprehensive understanding of CO photodissociation in astrophysical environments. As Visser et al. point out, line positions for the minor isotopologues 12C17O, 12C18O, 13C16O, and 13C18O are only tabulated for a subset of the CO bands between 91.2 and 118.8 nm. Very few isotopologue f-values have been measured, and those that have been reported in the literature are inconsistent—for example, Eidelsberg et al. (2006) report 12C16O and 13C16O f-values for the E(v = 1)–X(v = 0) band that differ by 16%, while Stark et al. (1992) report a 3% difference. Discrepancies of this size can have substantial effects on CO isotopic fractionation models (e.g., Lyons & Young 2005), wherein quite small differences in isotopologue absorption strengths can produce large fractionation signatures. At the time of the Visser et al. (2009) work, there were no published laboratory data on departures from standard Hönl–London intensity profiles (e.g., Morton & Noreau 1994) in any CO bands, so single f-values were reported for all bands. Yet, in the isoelectronic molecule N2, such departures are quite common (e.g., Stark et al. 2005, 2008; Heays et al. 2009). J-dependent effects are expected to be widespread throughout the CO spectrum, just as they are in N2; these nonstandard intensity distributions affect line-by-line self-shielding calculations, and they translate into temperature-dependent band f-values.
This report is part of a larger effort to catalog, interpret, and model the photoabsorption spectra of the isotopologues 12C16O, 12C17O, 12C18O, 13C16O, and 13C18O throughout the 91.2–111.8 nm region. The spectrum of CO in this region is characterized by a complex array of bands that display a wide range of strengths, linewidths, and rotational contours. At the longest wavelengths, transitions to the low-v vibrational levels of the first three Rydberg states (B1Σ+, C1Σ+, E1Π) in the series converging to the CO+ X2Σ+ ground state are prominent. Shortward of 100 nm, the spectrum becomes progressively more congested and complex as additional excited electronic states are accessed; the strongest transitions involve higher Rydberg states converging to the CO+ ground state and the CO+ A2Π state. Overviews and discussions of the spectroscopy of the 91.2–111.8 nm region are found in Huber (1997), Eidelsberg et al. (2004a), Lefebvre-Brion & Lewis (2007), Vázquez et al. (2009), and Lefebvre-Brion et al. (2010).
Here, we report line and band f-values for the six strong vibrational bands between 105.0 and 115.2 nm, associated with transitions from X(v'' = 0) to the v' = 0 and 1 levels of the B1Σ+, C1Σ+, and E1Π Rydberg states, in five CO isotopologues. We compute revised dissociation branching ratios for the C(v' = 0,1) and E(v' = 0,1) levels based on these f-values. This work complements our recent reports on f-values and dissociation rates of a number of bands in the 92.5–97.5 nm region in 12C16O (Eidelsberg et al. 2012) and 13C16O and 12C18O (Eidelsberg et al. 2014) as well as on term values of the A(v' = 0–9)–X(0) bands in 13C16O (Gavilan et al. 2013).
Direct astronomical observations of interstellar CO absorption into the low-v' levels of the B, C, and E states include those of the Copernicus satellite (e.g., Morton 1975; Snow 1975; Federman et al. 1980) and the FUSE satellite (e.g., Sheffer et al. 2003; Pan et al. 2005). Martian and Venusian atmospheric emissions originating from the B(0), B(1), C(0), and E(0) vibrational levels were recorded with the Hopkins Ultraviolet Telescope (Feldman et al. 2000), the FUSE satellite (Krasnopolsky & Feldman 2002) and the Cassini UVIS instrument (Gérard et al. 2011), and cometary B(0) and C(0) emissions were recorded with FUSE (Feldman et al. 2002).
Perhaps of broader significance than the direct astronomical observations, some of the bands studied in this report play central roles in CO photodissociation models. In particular, the E(0)–X(0) band, the strongest vibrational band in the entire CO spectrum after C(0)–X(0), is identified by van Dishoeck & Black (1988) and Visser et al. (2009) as being the most important contributor to the CO photodissociation rate in the outer regions of interstellar clouds, and the E(1)–X(0) band is a major contributor to isotopic fractionation effects (van Dishoeck & Black 1988). The theoretical understanding of the VUV absorption spectrum of CO is progressing, but it is not yet developed to the point where a predictive interpretation of the relevant predissociation mechanisms exists. The ab initio calculations of Cooper & Kirby (1987) and Kirby & Cooper (1989) established the importance of the D' 1Σ+ valence state (Wolk & Rich 1983) in the predissociation of levels in the 1Σ+ manifold. Tchang-Brillet et al. (1992) and Baker et al. (1995) developed a semi-empirical close-coupling model of the B1Σ+ and D' 1Σ+ states that reproduces trends in band positions and in predissociation linewidths for the low-v' levels of the B state, and Li et al. (1998) used ab initio potential curves and nonadiabatic couplings to study predissociation in both the B and C1Σ+ states. Recent work by Vázquez et al. (2009) Lefebvre-Brion et al. (2010), and Lefebvre-Brion & Eidelsberg (2012) has begun to extend the CO spectroscopic model to include the 1Π states. However, there is no model of the excited states of CO that is comparable to the comprehensive treatment of the Rydberg–Rydberg and Rydberg–valence interactions in N2 (Stahel et al. 1983; Spelsberg & Meyer 2001; Lewis et al. 2005a, 2005b, 2008) that successfully reproduces the predissociation and absorption strength patterns within the vibrational progressions of that molecule.
Laboratory studies of the low-v' B–X, C–X, and E–X bands are extensive. The principal spectroscopic results, including line positions, upper-state term values, and linewidths and upper-state lifetimes are reviewed and summarized in Visser et al. (2009). Two experimental approaches have been used in the determination of band f-values: optical absorption measurements (Letzelter et al. 1987; Eidelsberg & Rostas 1990; Eidelsberg et al. 2006; Stark et al. 1992, 1999; Federman et al. 2001; Sheffer et al. 2003) and inelastic electron-scattering measurements (Chan et al. 1993; Zhong et al. 1997). Optical absorption measurements provide, in principle, a direct determination of f-values. However, insufficient instrumental resolution can lead to optical depth effects, which, if not minimized or properly accounted for, result in significant systematic errors. The full-width at half-maximum (FWHM) Doppler width of 12C16O rotational lines at 295 K is 0.21 cm−1 at 110 nm. The majority of previous optical f-value measurements were of low or moderate resolution (ranging from ∼12 to ∼2 cm−1), with the exceptions of Stark et al. (1992, 1999), in which the E(0, 1)–X(0) and B(0, 1)–X(0) bands were measured with resolutions of 0.6 cm−1 and 0.14 cm−1, respectively. The measurements reported in this paper were carried out at the third-generation SOLEIL synchrotron facility in Saint Aubin, France. The vacuum ultraviolet Fourier transform spectrometer (VUV-FTS) on the DESIRS beam line, combining high spectral resolution (0.32 cm−1 or 0.22 cm−1 for the majority of the measurements) and a high signal-to-noise ratio (S/N), has allowed us to improve upon the accuracy and consistency of earlier results and to extend CO absorption measurements to include individual line strengths.
2. EXPERIMENTAL DETAILS
The VUV-FTS is a permanent end-station of the high-resolution absorption spectroscopy branch of the DESIRS beamline (Nahon et al. 2012) at Synchrotron SOLEIL. The beamline undulator provides a continuum background with a 7% bandwidth (6400 cm−1 FWHM, or 7.7 nm, at 110 nm). After a gas-filter chamber removes the unwanted high harmonics emitted by the undulator source, the continuum light passes through a gas-sample chamber before entering the VUV-FTS. Figure 1 displays a typical transmission spectrum.

Figure 1. DESIRS absorption spectrum of the 12C16O B(0)–X(0) and B(1)–X(0) bands. The feature at 91900 cm−1 is the C(0)–X(0) band.
Download figure:
Standard image High-resolution imageHigh-purity gases [12C16O (Alphagaz, 99.997%), 13C16O (Messer, 13C 99.1%; 16O 99.95%), 12C18O (ICON Isotopes, 18O 99%), 13C18O (Cambridge Isotopes, 13C 99%, 18O 95%), and a mixed 12C16O/12C17O/12C18O sample (ICON Isotopes, 12C16O 41.5%, 12C17O 48.5%, 12C18O 9.9%)] continuously flowed through a 10 cm long, 1.2 cm diameter, windowless absorption cell equipped with two 15 cm × 28 mm2 external capillaries. Two stages of differential pumping maintain the ultrahigh vacuum in the VUV-FTS and in the DESIRS beamline. The absorption cell pressure cannot be directly measured, nor is that pressure constant with position in the cell and capillaries. Pressures in the cell are estimated to have ranged from about 3 × 10−4 mbar to 5 × 10−3 mbar. A 0.1 mbar full-scale capacitance gauge monitored the CO pressure in the external gas-handling system. Small drifts in this pressure (⩽5%) were electronically monitored during absorption scans.
The VUV-FTS is described in detail in de Oliveira et al. (2009, 2011). It is a wave-front division interferometer and relies on a modified Fresnel bi-mirror configuration requiring only flat mirrors. The path difference is scanned through the translation of one reflector. This translation is measured by means of an external frequency-stabilized He–Ne laser, providing a very sensitive interferometric determination of the optical path difference variation. A post-recording computation performs a standard FTS phase correction and corrects for residual sampling errors. Owing to strict linearity in the wavenumber scale, the spectra can be put on an absolute scale using a single reference wavelength in the source. While the maximum resolution of the VUV-FTS is 0.08 cm−1, S/N considerations dictated a resolution of 0.32 cm−1 for most scans; a resolution of 0.22 cm−1 was employed for a subset of the measurements. Over the course of about 30 minutes of data collection, 50 co-added scans typically resulted in an S/N at the peak of the undulator bandpass of 150.
The broad and adjustable continuum bandpass of the DESIRS beamline allowed for the simultaneous recording of room-temperature absorption in pairs of CO bands, facilitating the determination of precise ratios of band strengths. For each isotopologue, three separate undulator settings were used to record: (1) the B(0)–X(0) and B(1)–X(0) bands (hereafter, for brevity, the B(0) and B(1) bands); (2) the C(0), E(0), and C(1) bands; and (3) the C(1) and E(1) bands. For each undulator setting, spectra were recorded with a range of column densities; care was taken to avoid optically thick features, and data analyses were generally restricted to features with peak optical depths less than ∼1.5 (absorption depths less than 77%). For a subset of the recorded spectra, external pressure readings were converted to absolute column densities by measuring absorption in the B(0) band. This required recording comparison spectra with different undulator bandpasses and identical gas pressures. The B(0) band f-value has been well-characterized by high-resolution (0.14 cm−1) laser-based measurements (Stark et al. 1999) and by the synchrotron-based measurements of Federman et al. (2001), and was adopted as a calibration standard with a band f-value of 0.0065(5). Calibrated column densities in the windowless cell ranged from 7.3 × 1013 to 2.5 × 1015 cm−2. Additional scans of the strong E(0) and C(0) bands were recorded at lower external pressures without calibration.
3. DATA ANALYSIS
With the exception of the E(1) band, all of the bands in this study are free of significant perturbations, and line assignments are straightforward. Initial line positions for the 12C16O, 12C18O, 13C16O, and 13C18O isotopologues were taken from the atlas of Eidelsberg et al. (1991). For the 12C17O isotopologue, line positions and molecular constants for the C(0) and C(1) bands were taken from Ubachs et al. (1995) and Cacciani et al. (2001), respectively, and E(0) molecular constants were taken from Cacciani et al. (1995). The strongly blended and perturbed Q branches of the E(1) band in all five isotopologues were spectroscopically analyzed by Ubachs et al. (2000), and their assignments were relied on in the initial assignment of line positions.
In the data reduction, the transmitted intensity, I(ν), where ν is the wavenumber, is related to the measured absorption cross section, σexp (ν), through application of the Beer–Lambert law,
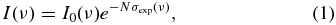
where σexp (ν) includes the effects of the finite instrument resolution, N is the column density of CO molecules, and I0(ν) is the background continuum level. For each line within a band, a least-squares fitting routine that accounts for the effect of the finite instrumental resolution was used to determine a value for the line's corrected integrated cross section. All rotational lines were modeled with Voigt profiles. The Gaussian component, calculated separately for each isotopologue and band, is the room-temperature Doppler width of ∼0.21 cm−1 FWHM. The onset of predissociation was established at J > 37 for the B(0) vibrational level of 12C16O and at J > 17 for the B(1) level (Eidelsberg et al. 1987). For the range of upper-state rotational levels sampled in our room temperature spectra, typically J ⩽ 25, there are no reports of measured line broadening in either vibrational level, and the Lorenztian component was set to zero for lines in the B(0) and B(1) bands for all isotopologues. Predissociation is known to occur for all rotational levels in the E(0), E(1), and C(1) states, but the associated line broadening is too small to be directly observed in our measurements. Published values of linewidths and lifetimes from laser-based measurements (Cacciani et al. 1998, 2001; Ubachs et al. 2000) were used to establish the Lorenztian components of lines in bands terminating in these states as well as in the C(0) band. The Lorentzian components ranged from 0.003 cm−1 FWHM for lines in the C(0) band of all isotopologues (Cacciani et al. 2001) to 0.036 cm−1 FWHM for the E(1) band of 13C18O (Ubachs et al. 2000).
The instrument function is described by a sinc function (0.32 cm−1 FWHM for most spectra), which results from the finite path difference in the recorded interferogram. In their study of the 14N15N spectrum with the DESIRS VUV-FTS, Heays et al. (2011) report the necessity of including an additional Gaussian component in the instrument function to account for fitted linewidths that were ∼0.05 cm−1 broader than expected. They attributed this to the nonideal collimation of the synchrotron beam entering the VUV-FTS. This correction was not adopted in the present study, as it has a much smaller effect on measured line strengths than on measured linewidths.
By varying the position and integrated cross section for each line, the least-squares fitting routine minimized the difference between a model transmission spectrum (calculated by a convolution of the line's Voigt profile in absorption with the instrumental sinc function) and the measured transmission spectrum. Figure 2 illustrates the quality of a fit to the B(0) band in 12C16O. Fitting uncertainties in the integrated cross sections for each line were assessed by the least-squares routine; they varied according to the S/N of each spectrum and the strength of the absorption, and were typically about 3% (one standard error). Uncertainties in the adopted literature values of linewidths produced very small additional uncertainties in the line cross sections, typically less than 1%.

Figure 2. Panel (a) Absorption spectrum of the 12C16O B(0)–X(0) band at an instrumental resolution of 0.32 cm−1. Panel (b) Expanded view of P-branch absorption (red) and least-squares fit (black). Note the presence of the instrumental sinc function oscillations in the data and in the fit. Fit residuals are shown in blue.
Download figure:
Standard image High-resolution imageUncertainties in the column densities of individual spectra were significantly larger than the statistical uncertainties of the fitting procedure. Because all absolute column densities were determined by absorption in the B(0) band (at the same external pressure settings), the 7% uncertainty in the B(0) f-value, adopted from combining the results of Stark et al. (1999) and Federman et al. (2001), applies to the derived column densities. Small variations in the external pressure during each 30 minute scan resulted in additional column density uncertainties, estimated to be less than 5%. Heays et al. (2011) discuss a further source of f-value uncertainty associated with the apparent effect of mechanical vibrations on the recorded spectrum—there is a small, but noticeable, variation in the center energy of the synchrotron-beam bandpass during sequential scans of the VUV-FTS scanning mirror. When multiple scans are summed (typically, 50 scans were summed to produce each spectrum), there are small distortions in line shapes and systematic errors can be introduced in line strengths. The line strength uncertainties are not yet well characterized, but they can be minimized by avoiding the analysis of deeply absorbed lines and the analysis of lines near the low-intensity wings of the synchrotron-beam bandpass. Importantly, mechanical vibration issues only negligibly affect the ratios of f-values of features appearing in the same spectrum.
4. RESULTS
The integrated cross sections of individual rotational lines, determined from the fitting procedure, were converted into line oscillator strengths according to (Morton & Noreau 1994)
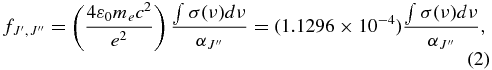
where the integrated cross section is in units of cm, and  is the fractional population of CO molecules in the J'' rotational level as determined from a normalized Boltzmann factor based on CO isotopologue ground state term values (Guelachvili et al. 1983). For transitions to unperturbed upper vibronic states, the rotational line f-values follow simple patterns described by Hönl–London factors (e.g., Morton & Noreau 1994), and the band f-value is related to the rotational line f-values by
is the fractional population of CO molecules in the J'' rotational level as determined from a normalized Boltzmann factor based on CO isotopologue ground state term values (Guelachvili et al. 1983). For transitions to unperturbed upper vibronic states, the rotational line f-values follow simple patterns described by Hönl–London factors (e.g., Morton & Noreau 1994), and the band f-value is related to the rotational line f-values by
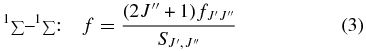
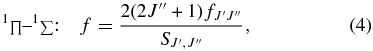
where the  are Hönl–London factors.
are Hönl–London factors.
For unperturbed bands, the band f-values derived from the application of Equations (3) and (4) to measured line f-values are independent of the rotational quantum number J. The B(0) and B(1) bands follow this description, and, hence, it is only necessary to specify a single band f-value to categorize the distribution of strengths among rotational lines. To simplify the least-squares fitting procedure for these bands, a single band f-value was varied and optimized, and Equations (3) and (4) were used to fix the ratios of line strengths in the fits.
The C(0), C(1), E(0), and E(1) bands in all isotopologues display weakly anomalous P-, Q-, and R-branch intensity patterns and are characterized by a J dependence of band f-values derived from Equations (3) and (4). For transitions from the ground state of CO, it is the upper-state wave function that is responsible for all departures from the standard line strength formulas. Accordingly, the band f-values derived from Equations (3) and (4) are considered as functions of J'. It was often sufficient to represent the J'-dependence of the derived band f-values as a linear function in J'(J' + 1); in such cases the P-, Q-, and R-branch intensity patterns were fit by varying and optimizing the J' = 0 intercept and the slope of the linear function. The J-dependent f-value patterns associated with specific bands are presented below.
4.1. B(0) and B(1) Bands
The B1Σ+ state is the lowest member of the nsσ Rydberg series (with n = 3) converging to the ground state of CO+, and it is the lowest-energy Rydberg state in the molecule. In 12C16O, the B(0) and B(1) bands are centered at 115.1 and 112.4 nm, respectively. Amiot et al. (1986) report weak, localized perturbations in the B(0) vibrational level at J = 4, 6 in 12C16O and at J = 18, 20 in 13C16O. We confirm the small energy shifts in 12C16O (≈0.02 cm−1), and we observe additional shifts, of similar magnitude, in lines terminating on J = 12 in 12C18O, J = 13 in 13C16O, and J = 17 in 13C18O. Drabbels et al. (1993) identify the perturbing state in 12C16O as the e3Σ− (v = 28) level. We find that the P- and R-branch intensity patterns in the B(0) bands of all five isotopologues studied are unaffected by the weak perturbations, and are adequately described by standard Hönl–London factors. And, as mentioned previously, the onset of predissociation in the higher-J levels of the B(1) state does not result in measurable broadening of the B(1) lines.
The previously measured f-value of the 12C16O B(0) band, 0.0065(5) (Stark et al. 1999; Federman et al. 2001), was adopted for all five isotopologues in the current study. The only known interaction affecting the B1Σ+ state is a strong homogeneous coupling to the weakly bound D' 1Σ+ valence state (Kirby & Cooper 1989; Tchang-Brillet et al. 1992). This coupling has been shown to reproduce the observed predissociation broadening of rotational levels in the B(2) vibrational level (Tchang-Brillet et al. 1992), and is responsible for the diffuse nature of the very weak higher-v' bands observed by Baker et al. (1995; Baker 2005) and Eidelsberg et al. (2004a). Although the B(0) level lies well below the crossing of the B and D' diabatic curves, isotopologue dependence in the B(0) f-value could, in principle, result from the coupling of these states due to the small shifts in the B(0) term values with nuclear reduced mass. To test this possibility, we used the parameters of the close-coupling model of the B1Σ+ and D' 1Σ+ states developed by Tchang-Brillet et al. (1992; Table II) to calculate the predicted changes in this f-value. We find that the model predicts a 1.7% difference in the B(0) band f-values of the lightest (12C16O) and heaviest (13C18O) isotopologues, with intermediate values for 12C17O, 13C16O, and 12C18O. These predicted f-value differences are significantly smaller than our experimental uncertainties.
Table 1 lists our measured ratios of the B(1) and B(0) f-values for four of the five isotopologues studied in this report. As described in Section 2, the DESIRS undulator bandpass (see Figure 1) allowed for the recording of absorption in both bands simultaneously. This eliminates all column density uncertainties in the determination of f-value ratios. The ratios are presented with uncertainties of 5%; this is a conservative estimate based on the fitting statistics for individual scans, the distribution of results from multiple scans of the same isotopologue, and possible systematics associated with small distortions in line strengths and line profiles introduced by mechanical vibrations in the beamline optics and the VUV-FTS (see Section 3).
Table 1. Measured Ratios of Band f-valuesa
| 12C16O | 12C17O | 12C18O | 13C16O | 13C18O | |
|---|---|---|---|---|---|
| B(1)/B(0) | 0.161(8) | b | 0.155(8) | 0.150(8) | 0.147(7) |
| C(0)/E(0) | 1.87(9) | 1.86(9) | 1.80(9) | 1.86(9) | 1.84(9) |
| C(1)/E(1) | 0.96(5) | 0.96(5) | 1.00(5) | 0.97(5) | 1.04(5) |
Notes. aUncertainties (in parentheses; 1 standard error) are in units of the last-quoted decimal place. bNo spectra available.
Download table as: ASCIITypeset image
Although the differences between the four B(1)/B(0) results are less than the stated uncertainties, there appears to be a weak trend in the f-value ratio, with the ratio decreasing with increasing nuclear reduced mass. Tchang-Brillet et al. (1992) predict a B(1)/B(0) f-value ratio of 0.15 for 12C16O. Application of their B–D' close-coupling model (the model parameters are independent of isotopologue) to the other CO isotopologues predicts an f-value ratio decrease of ∼3.7% from 12C16O to 13C18O. In this model, the B(1)–X(0) band gains strength from the intrinsically stronger D'–X transition (the D'–X transition moment is modeled to be ∼50 stronger than the B–X transition moment). The predicted f-value ratio decrease with increasing reduced mass can be understood as resulting from the lower energies of the heavier-isotopologue B(1) levels, moving them farther below the crossing of the B and D' potential curves and thus reducing their interactions with the D' state. Our measurements support this predicted trend, but the measurement uncertainties are too large for that support to be definitive.
4.2. C(0), C(1), E(0), and E(1) Bands
The C1Σ+ and E1Π states are the σ and π members of the 3p Rydberg complex associated with the CO+ X2Σ+ core. The C(0) and E(0) bands, at 108.8 and 107.6 nm in 12C16O, respectively, are the two strongest band features in the CO absorption spectrum; the C(1) and E(1) bands are located at 106.3 and 105.2 nm. Representative absorption spectra in 12C16O are displayed in Figure 3. As with the B(0) and B(1) bands, the DESIRS undulator bandwidth allowed for the determination in each isotopologue of ratios of the C(0) and E(0) f-values and, separately, the C(1) and E(1) f-values, without reference to absolute absorption column densities.

Figure 3. Representative absorption spectra of the C(0), C(1), E(0), and E(1) bands in 12C16O recorded at a resolution of 0.32 cm−1. All x-axis labels are wavenumbers in cm−1. The perturbation of the E(1) rotational levels by a crossing with the k3Π(v = 6) level causes a weakening of the intensities of the P(8) and R(6) lines and the appearance of two additional weak features (marked with vertical lines). Two molecular hydrogen lines (marked with stars) appear in the C(1)–X(0) spectrum.
Download figure:
Standard image High-resolution imageAll four bands display some level of J-dependence in their rotational branch intensity patterns; the J-dependences within each branch were modeled as linear functions in J'(J' + 1) with a common J' = 0 intercept. Because of the deep absorption present in the highly blended Q-branches of the E(0) and E(1) bands, the Q-branch line strengths were sometimes fit independently of the P- and R-branch patterns. The band f-values, determined by summing all of the individual line contributions, were generally insensitive to the particulars of the fitting procedure. A fit to a portion of the 12C16O E(0) band, including the congested Q-branch, is shown in Figure 4. The quality-of-fit of the assumed linear dependence with J'(J' + 1) of the measured f-values is imperfect. A more complex parameterization is not justified by the S/N of the measured spectra.

Figure 4. Portion of the 12C16O E(0)–X(0) absorption spectrum (black) showing the blended Q-branch and low-J lines in the R- and P-branches. The least-squares fit to the band (red) is offset for clarity. Fit residuals are in blue.
Download figure:
Standard image High-resolution imageThe J-dependences of the band f-values derived from Equation (4) for the P- and R-branch lines in the E(0) band of 12C16O are displayed in Figure 5, along with linear fits to a J'(J' + 1) dependence. The figure shows the P-branch lines increasing in strength and the R-branch lines decreasing in strength with J'(J' + 1). The opposite behavior is seen in the P- and R-branches of the C(0) band. This mutual line-intensity pattern in the two bands is expected as the result of an L-uncoupling interaction between the two components of the 3p Rydberg complex (Lefebvre-Brion & Field 2004, p. 394). That interaction, which is also responsible for the Λ-doubling in the E(0) level, has been evaluated by Hines et al. (1990) and by Haridass et al. (1994). Similar branch intensity patterns are seen in the E(1) and C(1) P- and R-branches in all isotopologues.

Figure 5. 12C16O E(0)–X(0) band f-values determined from measured P-branch (blue squares) and R-branch (red circles) rotational line f-values and Hönl–London factors. The J-dependences of the branch-specific f-values are represented by linear fits to J'(J' + 1) with a common J' = 0 intercept.
Download figure:
Standard image High-resolution imageOur measured f-value ratios for the two pairs of bands are listed in Table 1. We have assigned 5% uncertainties to these ratios. Within our estimated experimental uncertainties, there is no discernible dependence on the nuclear reduced mass in either ratio. In the absence of more definitive evidence, in the rest of this paper we adopt isotopologue-independent average values for the C(0)/E(0) and C(1)/E(1) ratios of 1.85(9) and 0.99(5), respectively.
All rotational levels in the E(1) vibrational state are predissociated by a currently unidentified mechanism, and there is an additional localized interaction with the rotational levels of the k3Π(v = 6) state (Ubachs et al. 2000) that causes line shifts and line broadening. Ubachs et al. (2000) measured line positions and linewidths in six CO isotopologues with a high-resolution laser system. They spectroscopically analyzed the highly blended Q-branches, identified multiple additional lines stemming from the E(1)–k(6) interaction, and modeled the interaction via a single J-independent spin–orbit coupling parameter. To successfully fit the Q-branch features in our spectra, wherein almost no individual lines are resolved, we adopted the spectroscopic parameters derived by Ubachs et al. (2000). A fit to a portion of the 13C16O E(1)–X(0) SOLEIL spectrum is shown in Figure 6. In our absorption spectra, we are able to identify and characterize a small number of perturbed lines not presented in the spectroscopic study of Ubachs et al. (2000). Level widths and term values for the upper states (when not reported by Ubachs et al.) of these lines are compiled in Table 2. For lines observed by Ubachs et al. (2000), our measured widths tend to be somewhat larger, though the results are consistent within the stated uncertainty ranges.

Figure 6. Portion of the 13C16O E(1)–X(0) absorption spectrum, showing the highly blended Q-branch. The Q(7) line is displaced to lower energy and broadened by the interaction of the E(1) level with the k3Π(v = 6) level (Ubachs et al. 2000). The Q(7) line of the k(6) – X(0) transition, which gains intensity from the interaction, is also identified. The least-squares fit to the band (red) is offset for clarity. Fit residuals are in blue.
Download figure:
Standard image High-resolution imageTable 2. Measured Widths and Term Values (T) of Perturbed E(1) Levelsa,b
| 12C16O | 13C16O | 12C18O | 13C18O | |
|---|---|---|---|---|
| this work c | this work c | this work | this work | |
| E(1) Je = 7d | 0.13(3) 0.10(1) | 0.15(3) 0.12 | 0.10(2) | |
| k(6) Je = 7 | 0.22(15) | 0.20(3) 0.15 | 0.22(15) | |
| E(1) Jf = 7 | 0.16(2) | |||
| k(6) Jf = 7 | 0.14(18) | 0.29(6) | ||
| T = 95131.95e | ||||
| k(6) Jf = 1 | 0.22(26) | |||
| T = 94985.14e |
Notes. aUncertainties (in parentheses; 1 standard error) are in units of the last-quoted decimal place. bAll results are FWHM in cm−1. cUbachs et al. (2000). dThe subscripts e and f designate the parities of rotational levels. eReferenced to J = 0 level of X(0); X(0) term values from Ubachs et al. (2000).
Download table as: ASCIITypeset image
4.3. Isotopologue Dependence of f-values
The extent of any isotopologue dependence of the C(v) and E(v) band f-values is of significance in the modeling of isotopic fractionation processes (see Section 1). Our most direct measures of these dependences come from the analyses of absorption spectra of mixed samples of CO isotopologues. For this purpose, we used a commercially prepared sample of 12C16O/12C17O/12C18O and a sample of 12C16O/12C18O prepared in the laboratory. The isotopologue concentrations in the samples were determined by a comparison of B(0) band absorption strengths. Under the assumption that the B(0) band f-value is independent of isotopologue (see our earlier discussion of this point), the sample concentrations were found to be 12C16O/12C17O/12C18O = 0.415(4)/0.485(5)/0.099(3) and 12C16O/12C18O = 0.293(6)/0.707(14), with uncertainties, based on fitting statistics, in units of the last-quoted decimal place. Ratios of f-values for the C(0), C(1), E(0), and E(1) bands in 12C16O and 12C17O, and in 12C16O and 12C18O, were determined from absorption spectra of the two mixed sample bottles at a resolution of 0.22 cm−1. The concentration ratio of ∼3:7 for the 12C16O/12C18O sample led to an optically thick Q-branch in the 12C18O E(0) band; the ratio of f-values for this band was determined from analysis of the P- and R-branches only. The 12C16O/12C18O E(1) f-value ratio was not determined because of excessive optical depth in the Q-branch of 12C18O and blending of lines in the P- and R-branches of the two isotopologues. Figure 7 shows a representative spectrum of the C(1) bands of the mixed 12C16O/12C17O/12C18O sample.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 7. Overlapping portions of the C(1)–X(0) bands in 12C17O and 12C16O, recorded from the mixed 12C16O/12C17O/12C18O gas sample at a resolution of 0.22 cm−1. Despite the overlapped bands, individual rotational lines in each isotopologue can be easily identified and analyzed.
Download figure:
Standard image High-resolution image{kind=link}
The measured ratios of band f-values from the mixed-gas samples are summarized in Table 3. The results support isotopologue-independent f-values for the C(v = 0,1) and E(v = 0,1) bands. Establishing isotopologue-specific band f-values to accuracies significantly better than 4% will require a more thorough experimental study of a full complement of mixed gas samples. In what follows, we provisionally adopt isotopologue-independent f-values for the C(v = 0,1) and E(v = 0,1) bands.
Table 3. Measured Ratios of Isotopologue-specific f-valuesa
| C(0)b | C(1) | E(0) | E(1) | |
|---|---|---|---|---|
| 12C17O/12C16O | 1.00(4) | 1.01(4) | 1.01(4) | 1.05(4) |
| 12C18O/12C16O | 1.01(4) | 1.00(4) |
Notes. aUncertainties (in parentheses; 1 standard error) are in units of the last-quoted decimal place. bX(0) is the lower state of all listed bands.
Download table as: ASCIITypeset image
4.4. Absolute f-values
The C(1) and E(1) band f-values were put on an absolute scale by recording the B(0) band, with its adopted f-value of 0.0065(5), at the same external pressure settings as used for the C(1) and E(1) bands. C(1) and E(1) f-values for all five isotopologues were initially determined in this way. The resulting uncertainties in the f-values include contributions from the uncertainty in the B(0) f-value, the statistics of the B(0), C(1), and E(1) band fits, possible variations in pressure over the course of the recording of sequential spectra, and possible systematics associated with mechanical vibrations of the VUV-FTS and the beamline optics. These uncertainties, added in quadrature, are estimated to be 10%. The resulting f-values—for C(1), ranging from 0.0032(3) in 12C17O to 0.0038(4) in 13C18O, and for E(1), ranging from 0.0033(3) in 12C17O to 0.0037(4) in 13C16O—are in fact consistent, within the stated uncertainties, with isotopologue-independent C(1) and E(1) f-values. We believe that the mixed-gas sample f-value ratios (Table 3) are more reliable than the range of C(1) and E(1) f-values found via the B(0) calibration procedure. We therefore adopt isotopologue-independent f-values, determined by a weighted average of the five measured f-values: for C(1)–0.00351(35), and for E(1)–0.00355(36). The final uncertainties, ∼10%, are dominated by the 7% uncertainty in the calibrating B(0) f-value.
Directly calibrating the C(0) and E(0) band f-values via B(0)–X(0) absorption features proved to be difficult owing to the large difference in f-values; the C(0) and E(0) f-values are about 20 and 10 times larger, respectively, than the B(0) band f-value. Instead, we relied on the C(1) band as a secondary standard for the C(0) and E(0) bands, as it appears weakly in the C(0) and E(0) scans. This approach minimized uncertainties associated with pressure drifts and mechanical vibrations. With an adopted C(1) f-value of 0.00351(35), we determine the isotopologue-independent C(0) and E(0) f-values to be 0.102(12) and 0.055(7), respectively. Uncertainties of 12% are assigned to these f-values because of the additional step of using the C(1) f-value as a secondary calibration standard.
Our absolute f-values are presented in Tables 4 and 5, which also include comparisons to experimental and theoretical literature values. Because of the J-dependence in the line strengths of the C(v = 0,1) and E(v = 0,1) bands, there will be some small temperature-dependence in the stated f-values (see Section 5). Our reported band f-values are thus only strictly valid at room temperature. For 12C16O, there is quite good consistency among the published optical absorption measurements for all six bands, with the exception of the early f-value survey of Letzelter et al. (1987), the results of which are consistently low, likely due to the difficulty of avoiding systematic optical-depth effects in low-resolution measurements. Aside from the Letzelter et al. (1987) f-values, there is only one measurement, that of the B(1)–X(0) f-value of Federman et al. (2001), that is marginally outside of the uncertainty limits of the other published f-values. The f-values of the electron energy-loss measurements of Chan et al. (1993) and Zhong et al. (1997) are more significantly scattered relative to our results than other optical measurements, but they are broadly consistent with the optical measurements. It should be noted that the uncertainties in our absolute f-values in Table 4 include the contribution of the 7% uncertainty in the calibrating B(0) band. Our measured ratios of pairs of band f-values are significantly less uncertain (Table 1).
Table 4. 12C16O Band f-valuesa,b
| B(0)c | B(1) | C(0) | C(1) | E(0) | E(1) | ||
|---|---|---|---|---|---|---|---|
| Optical Absorption | Res.d | ||||||
| This worke,f | 0.32 | 6.5(5)g | 1.05(12) | 102(12) | 3.51(35) | 55(7) | 3.55(36) |
| Letzelter et al. 1987 | 12 | 4.5(5) | 0.70(7) | 62(6) | 2.8(3) | 37(4) | 2.5(3) |
| Stark et al. 1992 | 0.6 | 49(5) | 3.0(3) | ||||
| Stark et al. 1999 | 0.14 | 6.5(6) | 1.1(1) | ||||
| Federman et al. 2001 | 9 | 6.7(7) | 0.80(12) | 123(16) | 3.0(4) | 68(7) | |
| Sheffer et al. 2003 | 4.5 | 1.06(11) | 3.3(11) | ||||
| Eidelsberg et al. 2006 | 2.4 | 3.6(3) | |||||
| Electron Energy Loss | |||||||
| Chan et al. 1993 | 390 | 8.0(4) | 1.32(7) | 118(6) | 3.56(18) | 71(4) | 3.53(18) |
| Zhong et al. 1997 | 40 | 5.98(93) | 114(14) | 3.22(94) | 64(8) | 4.67(66) | |
| Calculations | |||||||
| Kirby & Cooper 1989 | 2.1 | 0.3 | 118 | 1.8 | 49 | 5.0 | |
| Chantranupong et al. 1992 | 5.1 | 0.52 | 65 | 4.9 | 27 | 3.3 | |
| Rocha et al. 1998 | 4.8 | 0.43 | 89 | 2.9 | 49 | 5.0 |
Notes. aUncertainties (in parentheses; 1 standard error) are in units of the last-quoted decimal place. bf-values in units of 10−3. cX(0) is the lower state of all listed bands. dResolution in cm−1. eListed uncertainties include 7% uncertainty in B(0) f-value. See Table 1 for ratios of band f-values, which have smaller fractional uncertainties. fC–X and E–X band f-values strictly valid only at room temperature. gAdopted for column density calibrations from Stark et al. (1999) and Federman et al. (2001).
Download table as: ASCIITypeset image
Table 5. CO Isotopologue Band f-values: Comparisons to Literaturea,b
| This workc | Eidelsberg & Rostas 1990 | Stark et al. 1992 | Eidelsberg et al. 2006 | |
|---|---|---|---|---|
| Resolutiond | 0.32 | 12 | 0.6 | 2.4 |
| 13C16O | ||||
| B(0)e | 6.5(5)f | 4.5(5) | ||
| B(1) | 0.98(12) | 0.72(7) | ||
| E(0) | 55(7) | 37(4) | ||
| C(1) | 3.51(35) | 2.70(27) | ||
| E(1) | 3.55(36) | 2.42(24) | 3.1(3) | 4.2(5) |
| 13C18O | ||||
| E(1) | 3.55(36) | 3.5(1) |
Notes. aUncertainties (in parentheses; 1 standard error) are in units of the last-quoted decimal place. bf-values in units of 10−3. cThe only listed isotopologue f-values are those that can be compared to literature values. With the exception of the B(1) band, all f-values reported in this work are isotopologue-independent. B(1) isotopologue f-values can be calculated from ratios in Table 1. dResolution in cm−1. eX(0) is the lower state of all listed bands. 12 C16O f-value adopted from Stark et al. (1999) and Federman et al. (2001); isotopologue independence of this f-value is discussed in the text.
Download table as: ASCIITypeset image
Prior to our work, 13C16O and 13C18O f-value measurements of selected bands were reported, and are listed in Table 5. Our measured ratios of isotopologue-specific f-values in 12C16O, 12C17O, and 12C18O for the C(v = 0,1) and E(v = 0,1) bands (Table 3) strongly point to isotopologue independence of these f-values at the 5% level. The 13C16O isotopologue has a nuclear reduced mass that is almost identical to that of 12C18O; for this reason we do not expect any isotopologue-dependence to appear in the 13C16O f-values. 13C18O has a larger reduced mass than 12C16O, 12C17O, or 12C18O, so we cannot rule out some isotopologue dependence in the 13C18O f-values. In their study of CO photodissociation, Visser et al. (2009) adopted isotopologue-independent f-values for the E(0) and C(1) bands, but isotopologue-dependent f-values for the E(1) band that vary by 20% based on the measurements of Eidelsberg et al. (2006). While the Eidelsberg et al. (2006) E(1) f-values are consistent with our present results when considering the combined uncertainties, our present results do not support an E(1) f-value isotopologue dependence.
4.5. Photodissociation Branching Ratios
Table 6 lists calculated photodissociation branching ratios (also referred to as predissociation probabilities) for the C(v = 0,1) and E(v = 0,1) vibrational levels. The branching ratios are derived from lifetime (Cacciani et al. 1998, 2001) and linewidth (Ubachs et al. 2000) measurements combined with our reported f-values. The methodology of the calculation is fully described in Cacciani et al. (1998). Briefly, the dissociation branching ratio, η, is determined by the lifetime, τ, and the radiative decay rate, Arad, via

Table 6. Dissociation Branching Ratiosa
| 12C16O | 12C17O | 12C18O | 13C16O | 13C18O | |
|---|---|---|---|---|---|
| C(0) | |||||
| this workb,c | −0.07(17) | −0.07(17) | 0.10(15) | ||
| Cacciani et al 2001 | 0 | 0 | 0.17 | ||
| C(1) | |||||
| this work | 0.62(8) | 0.83(3) | 0.87(2) | 0.82(3) | 0.91(2) |
| Cacciani et al 2001 | 0.65 | 0.84 | 0.88 | 0.83 | 0.91 |
| E(0) | |||||
| this work | 0.83(3) | 0.75(5) | 0.77(5) | ||
| Cacciani et al 1998 | 0.78–0.85 | 0.69–0.84 | 0.69–0.84 | ||
| E(1) | |||||
| this work | 0.971(5) | 0.960(9) | 0.960(7) | 0.963(7) | 0.945(12) |
| Cacciani et al 1998 | 0.972–0.986 | 0.953–0.976 | 0.936–0.956 | ||
| & Ubachs et al 2000 |
Notes. aCalculated from band f-values combined with lifetime and linewidth measurements (see the text). bUncertainties (in parentheses; 1 standard error) are in units of the last-quoted decimal place. cBlank entries indicate that no lifetime or linewidth data are available.
Download table as: ASCIITypeset image
Measured linewidths (Ubachs et al. 2000) are converted to lifetimes by

where Γ is the natural linewidth (FWHM) and c is the speed of light. Arad is the sum of Einstein-A coefficients for all radiative transitions from the upper state. An Einstein-A coefficient for a single vibrational band, e.g., E(0)–X(0), is directly determined from the band f-value (Morton & Noreau 1994), and in each of the cases considered here, dominates the sum. Using radiative branching ratios estimated from band f-values within the E–X system and from calculated branching into the E–A and E–B systems (Kirby & Cooper 1989), Cacciani et al. (1998) estimate that Arad is 16% larger than the A coefficient for the E(0) band. Cacciani et al. (1998) identified the f-values of the E(0) and E(1) bands as the major contributors to the uncertainty in the dissociation branching ratios for E(0) and E(1), and they presented ranges of branching ratios based on the existing f-value literature (Eidelsberg & Rostas 1990; Stark et al. 1992; Chan et al. 1993). In Table 6, we adopted the relevant estimates of Cacciani et al. (1998) along with our newly measured E(0) and E(1) band f-values to derive a set of branching ratios. For the 12C17O and 12C18O isotopologues, we used the E(1)–X(0) linewidth measurements of Ubachs et al. (2000) to establish the upper state lifetimes. Fractionation effects should be small due to any differences in isotopologue branching ratios for the E(1) level, as all of the branching ratios are very close to unity. This is not the case for the E(0) level, and either linewidth or lifetime measurements are needed to establish the E(0) branching ratios for the 12C17O and 12C18O isotopologues.
There is presently no experimental evidence for predissociation in the C(0) vibrational level of 12C16O. Cacciani et al. (2001) assumed that their measured lifetime for the C(0) level in 12C16O and 13C16O, 1.78 ns, represents a purely radiative decay, and they attributed the shorter lifetime for the 13C18O isotopologue (1.50 ns) to a dissociative contribution to the decay (branching ratio of 17%). We did not make these assumptions, but rather used our measured band f-values and the lifetime measurements of Cacciani et al. (2001) to derive the dissociation branching ratios. Our results in Table 6 corroborate the conclusion that there is no predissociation in the 12C16O and 13C16O C(0) levels, and they are in good agreement with earlier determinations for the E(0), E(1), and C(1) levels.
5. DISCUSSION
The work presented here is part of a larger effort to establish a reliable high-resolution database for CO isotopologue absorption features in the 91.2–111.8 nm region (Eidelsberg et al. 2012, 2014). Our 12C16O f-values for the B–X, C–X, and E–X bands are directly applicable to the determination of column densities in diffuse molecular clouds (e.g., Sheffer et al. 2003; Crenny & Federman 2004). In this context, the moderate branch-dependences and J-dependences in the C–X and E–X bands (and the absence of any such dependences in the B–X bands) allow for the determination of temperature-dependent band f-values that can be applied to the interpretation of astrophysical observations. The resulting temperature dependences are, in fact, very modest. We calculate that the T = 50 K 12C16O C(0) and C(1) band f-values are 1.4% and 0.8% smaller, respectively, than the room-temperature f-values, and T = 10 K f-values are 1.6% and 1.1% smaller than the respective room-temperature f-values. For the E(0) and E(1) bands, the percentage changes are +3.0% and −1.7% at T = 50 K, and +3.7% and −4.2% at T = 10 K. We caution against extrapolating our results to temperatures above 300 K, as branch-dependences and J-dependences can change radically due to perturbations of high-J levels.
In addition to providing improved f-values for the interpretation of astronomical observations, our measurements will inform ongoing efforts to develop comprehensive treatments of the dissociation mechanisms in CO (e.g., Lefebvre-Brion et al. 2010; Lefebvre-Brion & Eidelsberg 2012). Any successful quantitative model of CO dissociation mechanisms must reproduce the isotopologue-specific patterns of absorption strengths and line widths (or the isotopologue independence of these patterns). Our most precise measurements come from analyses of absorption in mixed gas samples of 12C16O/12C17O and 12C16O/12C18O. These measurements show minimal or no isotopologue dependence in the C(v = 0,1) and E(v = 0,1) band f-values at the ∼5% uncertainty level. The absence of measureable isotopologue dependence in our band f-values, as well constraining CO dissociation models, is an important factor in establishing more reliable photodissociation branching ratios.
Recent CO photolysis experiments performed at the Advanced Light Source (ALS) synchrotron (Chakraborty et al. 2008, 2012) reported unexpectedly large enrichments in 17O in the CO2 product for the synchrotron beam centered at 105.17 and 107.6 nm. Chakraborty et al. concluded that the wavelength dependence of the relative enrichments in 17O and 18O do not support the CO self-shielding theory for the origin of the nearly equal depletions seen in 17O and 18O in CAIs (Clayton 2002; Yurimoto & Kuramoto 2004; Lyons & Young 2005), which would have implications for the UV environment experienced by the solar system. Understanding the full significance of the distribution of oxygen isotopes in CAIs for solar system formation requires an accurate assessment of isotope fractionation in all of the astrochemically relevant dissociation bands of CO, including the long-wavelength bands reported in this manuscript.
CO absorption in the ALS bandpasses at 105.17 and 107.6 nm is dominated by the C–X and E–X bands presently reported. Chakraborty et al. attributed the 17O enrichment to "accidental predissociation" in the E(1) band, due to interaction with the k3Π(v = 6) excited state, where an isotopologue-dependent alignment of several rotational levels occurs. Such near-resonance spin–orbit coupling between triplet and singlet states is known to produce strong isotope effects in some molecules (see discussion in Chakraborty et al. 2008), but in this case the interaction is very weak (Ubachs et al. 2000) and is unlikely to be the source of the measured 17O enrichment (Lyons et al. 2009; Federman & Young 2009; Yin et al. 2009). The minimal isotopologue dependence of our reported C–X and E–X f-values places a further strong constraint on the interpretation of the large 17O enrichments in the photolysis experiments: f-value variations can be ruled out as being responsible for the large 17O enrichments.
Self-shielding by C18O is a more likely explanation. Using our measured band f-values and J-dependent line strengths along with line widths reported by Ubachs et al. (2000), we developed synthetic room-temperature cross section profiles for the E(1) band for each isotopologue. At the lowest photocell column density reported by Chakraborty et al. (2012) for the ALS bandpass centered on the E(1) band at 105.17 nm–2.6 × 1017 cm−2—the peak optical depths of the strong Q-branches in 12C16O, 12C17O, and 12C18O are ∼570, 0.22, and 1.2, respectively. With these peak optical depths, absorption in the 12C18O Q-branch, which normally accounts for ∼50% of the E(1) band absorption, will be significantly reduced by self-shielding while absorption in the 12C17O Q-branch will be only moderately affected by self-shielding. Hence, the 17O/18O ratio in the photolysis products should be enhanced. For the ALS measurements centered on the E(0) band at 107.61 nm, the peak E(0) Q-branch optical depths for the lowest photocell column density (1.3 × 1017 cm−2) are ∼1200, 0.46, and 2.5 for 12C16O, 12C17O, and 12C18O. Again, significant self-shielding should be expected in 12C18O relative to 12C17O. Aside from self-shielding effects, there remains the possibility that isotope-dependent dissociation probabilities for C17O and C18O could account for the 17O enhancement in the photolysis products (Lyons 2014). The dissociation branching ratios derived from our f-value measurements and literature line widths (Table 6) should help clarify this possible contribution. In the future, we plan to use the f-values reported in this paper to more quantitatively evaluate the effects of self-shielding on the fractionation results of Chakraborty et al.
This research was supported by funds from NASA (grants NNX09AC5GG to Wellesley College and NNG 06-GG70G and NNX10AD80G to the Univ. of Toledo), CNRS (France), and Programme National Physico-Chimie du Milieu Interstellaire (PCMI). ANH was supported by grant number 648.000.002 by the Netherlands Organization for Scientific Research (NWO) via the Dutch Astrochemistry Network. L.G. and J.L.L. acknowledge the financial support of the European Community 7th Framework Programme (FP7/2007–2013) Marie Curie ITN under grant agreement # 238258 ("LASSIE" contract).