
Abstract
Nano iron oxo-hydroxides have numerous and increasing applications in biology and medicine. Assessment of their uptake and toxicity often requires cell culture but maintaining iron oxides uniformly nano-dispersed in such conditions can be challenging. We describe a balanced salt solution (BSS) compatible with cellular assays for use in such investigations. We determined hydrodynamic particle size and dispersibility of nano iron in BSS. The BSS, containing 130 mM NaCl, 10 mM KCl, 1 mM MgSO4, 5 mM glucose and 1.8 mM CaCl2 in 10 mM PIPES buffer (pH 7.4), was capable of maintaining nanoparticulate tartrate-modified Fe(III) oxo-hydroxide (i.e. nano Fe) mono-disperse (≥95% nanoparticulate) with a mean hydrodynamic particle diameter of 6.1 ± 0.3 nm. This size was similar to the native form of the nano Fe material (i.e. as synthesized) with a mean hydrodynamic particle diameter of 4.3 ± 0.1 nm in water. Furthermore, we show that BSS also adequately maintains nano Fe dispersion when supplemented with inhibitors of particle uptake or lysosomal acidification, namely chloropromazine and monensin, and when used at pHs 6.5 or 5.8. In conclusion, we provide a method for nanodispersion of iron oxo-hydroxides that is suitable for short term (1–3 h) cellular exposure investigations.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Research and development of novel iron oxo-hydroxide nanomaterials is fast growing [1–10] and, although nano iron oxo-hydroxide structures are not a new phenomenon, it is now possible to characterize them much better than before due to marked technological advances, for example with electron microscopy. Engineered nanoparticulate iron oxides and hydroxides are amongst the most widely used nanomaterials in biomedical and food applications due to their safety and versatility [5, 11]. For example, super paramagnetic iron oxides (SPIOs) with primary particle diameters over 26 nm have been used for some decades as contrast agents for MRI [9, 12, 13]. Nano iron oxo-hydroxides are also used in intravenous iron replacement therapy for iron deficiency anaemia, typically coated with a carbohydrate shell and with a hydrated size range of 8–30 nm (although primary particle diameter of the iron oxo-hydroxide core may be as small as 1–3 nm) [14, 15]. Applications as food fortificants and dietary supplements have recently gained interest [16, 17]. Hilty et al reported the development of iron and zinc nanostructured phosphates/oxo-hydroxides with diameters below 10 nm for use as iron fortificants. These can be added to foods due to their highly compatible organoleptic properties [16]. Small nanoparticles of ferric iron oxo-hydroxides coated with hemin (primary particle diameter 4.5 nm) or polymaltose (primary particle diameter 2.5 nm), or doped with organic acids (primary particle diameter ∼2 nm) are also used or under investigation as oral iron supplements [14, 18–20].
Physico-chemical properties of nanomaterials, in the environment used for their cellular testing, will influence their physical behaviour (e.g. agglomeration or aggregation), cell uptake and toxicity [21, 22]. Non-coated iron oxo-hydroxide nanoparticles have reactive surfaces and, on one hand, readily self-associate leading to precipitation from suspension. On the other hand, reactive iron oxo-hydroxides may dissolve in aqueous solutions if exposed to different chemical components (such as serum components that chelate metals) or pH conditions. Coated iron oxides, such as SPIOs, are generally protected from agglomeration in media due to the stabilization imparted by their carbohydrate coatings.
In the environment and in biological systems particles acquire a (protein) corona, whereby soluble molecules are strongly adsorbed to the particle surface [23, 24]. This undoubtedly modifies the particle properties and adds a layer of complexity in translating between observations in culture of particle–cell interactions and the in vivo situation. Furthermore, mimicking in vitro, for example, the corona imparted during gastrointestinal digestion for orally ingested nanoparticles, would be extremely difficult and complex because proteins are partly digested in the upper gastrointestinal tract. However, ensuring that these particles remain disperse, so that at least cell interactions are studied for the particle itself and not agglomerates or aggregates, should be possible. It is also important and relevant physiologically to examine in in vitro assays the cellular effects of the 'naked' particles since, once particles are taken up, the protein corona would be digested in cellular lysosomes and cells would be exposed to the 'naked' particle at some stage of the interaction with particles.
Therefore, here, we have developed a medium that is suitable for cells in culture and that is able to maintain non-coated ferric iron (Fe(III)) nanoparticles genuinely disperse at peri-neutral pH conditions. We anticipate that the strategy for optimization of the medium presented here could be applied to other mineral nanoparticles (e.g. Zn, Cu, or Al based) when serum-free cellular assays are required to assess particle–cell interactions.
2. Experimental
2.1. Materials
All chemicals and reagents were obtained from Sigma-Aldrich (Dorset, UK) unless otherwise stated. All material suspensions were prepared in 18.2 MΩcm−1 ultrapure water (H2OUHP).
2.2. Preparation of nano Fe(III)
The nanoparticulate tartrate-modified Fe(III) poly oxo-hydroxide material (herein referred to as nano Fe) is a proposed novel iron supplement [20] and was produced following the protocol described by Powell et al [19]. Briefly, a solution containing tartaric acid for particle modification, and adipic acid used as a buffer [19], was mixed with an acidified solution of Fe(III) chloride to achieve resulting final molar ratios of 1:1:2 (tartaric acid: adipic acid: Fe) and [Fe] = 40 mM. This suspension was then titrated to pH 7.4 using NaOH. This nano iron suspension was prepared fresh for each experiment.
2.3. Media
Unless otherwise stated, the nano Fe material was dispersed into different aqueous media at pH 7.4 and at physiologically relevant Fe concentrations (200–1000 μM) and the phase distribution and hydrodynamic size were determined as described below. The different aqueous media used individually or combined were: minimum essential medium (MEM; PAA Laboratories, Yeovil, UK); H2OUHP (ultra-high purity); 1 mM NaH2PO4; 1.8 mM CaCl2; 10 mM PIPES (piperazine-N,N'-bis(2-ethanesulfonic acid)). The MEM contained (mM): L-arginine hydrochloride (0.6), L-cystine (0.1), L-glutamine (2), L-histidine hydrochloride (0.2), L-isoleucine (0.4), L-leucine (0.4), L-lisine hydrochloride (0.4), L-methionine (0.1), L-phenylalanine (0.2), L-threonine (0.4), L-tryptophan (0.05), L-tyrosine (0.2), L-valine (0.4), choline chloride (0.007), D-calcium pantothenate (0.002), folic acid (0.002), niacinamide (0.008), pyridoxal hydrochloride (0.005), riboflavin (0.0003), thiamine hydrochloride (0.003), i-inositol (0.01), CaCl2 (1.8), MgSO4 (0.8), KCl (5.3), NaHCO3 (26), NaCl (117), NaH2PO4 (1), D-glucose (5.6), phenol red (0.03). Additionally, a balanced salt solution (BSS) containing 130 mM NaCl, 10 mM KCl, 1 mM MgSO4, 5 mM glucose and 1.8 mM CaCl2 in 10 mM PIPES buffer, at pH 7.4 was used. The BSS was also used when supplemented with various chemical inhibitors/stimulants of cellular uptake pathways: chloroquine (100 μM), Mas-7 (5 and 20 μM), CoCl2 (4, 10 and 20 mM), chlorpromazine (100 μM) and monensin (30 μM).
2.4. Physicochemical characterization of nano Fe(III)
2.4.1. Phase distribution
Fractionation of the Fe into percentages of microparticulate, nanoparticulate and soluble Fe, for each of the iron-supplemented media was carried out with centrifugation and ultrafiltration. First these were centrifuged (10 000g, 5 min) and the sediment taken to be the microparticulate Fe fraction. In order to isolate the soluble Fe, and to distinguish it from nanoparticulate Fe, the supernatant was further ultrafiltered (3,000 Da MWCO; 10 000g, 10 min). The Fe content of all fractions (total, supernatant and ultrafiltrate) was determined by inductively-coupled plasma optical emission spectrometry (ICP-OES JY 2000, Horiba Jobin Yvon, Stanmore, UK) at 259.94 nm. Each measurement was performed in triplicate. ICP-OES standards and samples were diluted in 0.5% HNO3 to concentrations in the range 0–1000 ppb. The Fe content in these fractions was expressed as percentage ± SD in relation to total Fe content as follows:
![$\begin{array}{lllllllllllllll} \left[ (\%)\;{\rm Fe}\;{\rm microparticulate} \right]=\left[ \left( {\rm total}\;{\rm Fe}-{\rm Fe}\;{\rm supernatant} \right)/{\rm total}\;{\rm Fe} \right]\times 100, \\ \left[ (\%)\;{\rm Fe}\;{\rm nonoparticulate} \right]=\left[ \left( {\rm Fe}\;{\rm supernatant}-{\rm Fe}\;{\rm ultrafiltrate} \right) \right./\left. {\rm total}\;{\rm Fe} \right]\times 100, \\ \left[ \left( \% \right)\;{\rm Fe}\;{\rm soluble} \right]=\left[ \left( {\rm Fe}\;{\rm ultrafiltrate} \right)/ \right.\left. {\rm total}\;{\rm Fe} \right]\times 100. \\ \end{array}$](https://content.cld.iop.org/journals/2053-1591/2/1/015403/revision1/mrx507402ieqn1.gif)
2.4.2. Particle size
The hydrodynamic particle size of the nano fraction was determined by dynamic light scattering (DLS) using a Zetasizer, Nanoseries Nano ZS (Malvern Instruments, Worcestershire, UK). DLS was used to measure the size of the particles in suspension in the different test media immediately after centrifugation to remove any sedimenting large agglomerates that would interfere with the measurement (10 000g, 5 min). The minimum particle concentration that provided valid DLS data corresponded to [Fe] = 500 μM. Each measurement was performed in triplicate. Dry primary particle size was determined by transmission electron microscopy (TEM) using a Philips CM200 FEG-TEM operating at 197 keV and fitted with a Gatan Imaging Filter (GIF 200) and Oxford Instruments UTW ISIS x-ray detector (EDS). Samples were prepared for TEM by drop-casting the nano iron 'as synthesized' suspension on holey carbon support film (Agar Scientific) and leaving to air dry.
2.4.3. Size stability
The nano Fe material was thoroughly dispersed in MEM or BSS at 500 μM Fe concentration and aliquots for size and phase distribution measurements were taken at different time-points over 24 h.
2.5. Data analysis
Unless stated otherwise, results are presented as means with standard deviations (SD). DLS results were calculated with the Malvern Zetasizer Nanoseries software (Nano ZS, Malvern Instruments, Worcestershire, UK) and are reported in volume as mean particle diameter, dv0.9 (diameter where 90 per cent of the population stays below), and dv0.1 (diameter where 10 per cent of the population stays below). One-way ANOVA with Sidak post-hoc test for multiple comparisons was used to statistically compare mean diameter at different time points and with different media (level of significance set to p < 0.05). All statistical analysis were performed using GraphPad Prism version 6 for Windows (GraphPad Software, San Diego, California, USA).
3. Results and discussion
Most mineral nanoparticles will readily agglomerate in serum free tissue culture medium [22, 25, 26] and, therefore, a thorough characterization of the nanomaterial from synthesis through to 'as used' in cellular or in vivo studies is vital. Here, immediately following synthesis, the phase distribution profile of the nano Fe showed that 87 ± 1% of the total iron was present in the nanoparticulate fraction (figure 1(A)). The mean hydrodynamic particle diameter as measured by DLS in the synthesis solution was 4.3 ± 0.1 nm (n = 3, PDI range 0.215–0.222) and a typical DLS measurement is shown in figure 1(B). The dry primary particle size of the nano Fe material, as determined by TEM, was 2–3 nm (figure 1(C)), consistent with a hydrodynamic size being 1.5–2 nm greater. When nano Fe was dispersed in a commonly used serum-free tissue culture medium (MEM), at Fe concentrations that are used in cellular uptake studies of supplemental iron [27, 28], the particles agglomerated/aggregated almost completely with 97 ± 2% of the Fe present in the microparticulate fraction (figure 2(A)). Even if the very small fraction that didn't sediment with centrifugation (2.3 ± 1.7%) suspended particles of 321 ± 105 nm in diameter were observed after 30 min with no significant changes in their size over a 24 h period (figure 2(B)).
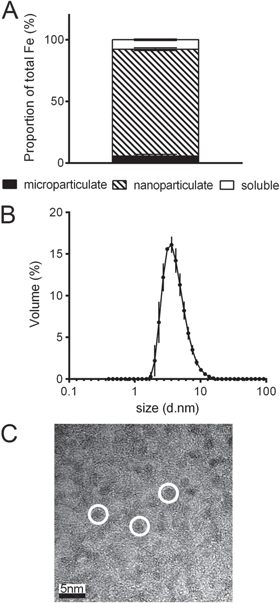
Figure 1. Particle size and phase distribution of nano Fe(III) oxo-hydroxide ('nano Fe') 'as synthesized'. (A) Phase distribution of the nano Fe material following synthesis, i.e. fractional percentage of microparticulate (black), nanoparticulate (hatched) and soluble Fe (white). Values are mean ±SD of three independent experiments. (B) Hydrodynamic particle size distribution of nano Fe following synthesis as measured by dynamic light scattering (DLS). Values are expressed as mean diameter ±SD (n = 3 independent measurements) on a log10 scale. (C) Transmission electron microscopy (TEM) images collected from a drop of the nano Fe suspension following synthesis showing non-aquated (dry) primary particle sizes of 2–3 nm (example particles are shown in white circles). Scale bar is 5 nm. Detailed methodology is available in the methods section.
Download figure:
Standard image High-resolution image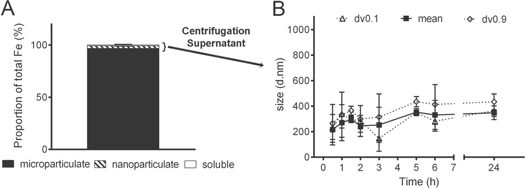
Figure 2. Particle size of nano Fe(III) oxo-hydroxide ('nano Fe') in serum-free tissue culture media (MEM). (A) Phase distribution of the material dispersed in MEM at an Fe concentration of 500 μM. Data presented for the fractional percentage of microparticulate (black), nanoparticulate (hatched) and soluble Fe (white). Samples were collected immediately following mixing in MEM. Values are mean ±SD (n = 3 independent experiments). (B) Hydrodynamic diameter (DLS) of the material in the small fraction (2.3 ± 1.7%) that didn't sediment with centrifugation (10 000g, 5 min) as per panel (A). The suspension was allowed to equilibrate for 30 min. Data are reported in volume as mean (±SD) particle diameter with dv0.9 (±SD) and dv0.1 (±SD), n = 2 independent measurements. Differences in mean diameter over time are not statistically significant (p ≥ 0.05). Detailed methodology is available in the methods section.
Download figure:
Standard image High-resolution imageBecause the nano Fe material might be completely agglomerated/aggregated in MEM, we next considered which components of MEM were mostly responsible. Fe(III) oxides bind phosphate with high affinity [29–31] with typically two hydroxyl groups or water molecules being replaced by a phosphate anion resulting in a stable bidentate complex [32, 33]. However, when calcium is also present in the medium, bridging coagulation/flocculation can occur which leads to formation of large polymers [34–36]. Indeed, when the dried nano Fe material was re-dispersed in an aqueous solution containing calcium and phosphate together at the same level as in MEM the nano Fe significantly agglomerated/aggregated with a mean diameter of 470 ± 139 nm (p < 0.0001, figure 3(A)). However, the material remained in the small nanoscale range, i.e. close to size as synthesized, when re-dispersed in water alone, in water plus phosphate alone and in water plus calcium ions alone (figure 3(A)). Since Ca ions are indispensable for many cellular functions, and in particular for ion and particle uptake mechanisms [37–39], they cannot be removed from the media used for cellular uptake assays and, therefore, we continued development with dissolved Ca ions, in the absence of phosphate, as our simplified cell culture medium. PIPES buffer (pKa = 6.8 at 25° C, [40]) did not induce significant agglomeration/aggregation of the nano Fe material, even in the presence of calcium (figure 3(A)), because this agglomeration is not driven by pH but by bridging coagulation/flocculation of iron oxides with adsorbed phosphate in the presence of calcium as detailed above. Therefore, PIPES was the buffer selected for physiological pH investigations and for the basis of our optimized BSS. All other conventional BSSs used in cell culture, namely Hank's balanced salt solution or Earle's balanced salt solution are based on phosphate buffers and, therefore, would also lead to the agglomeration of the particles in the presence of calcium as explained above. The final BSS contained additional nutrients and salts required for normal cellular function and to maintain ionic strength, such as glucose, sodium, potassium and magnesium, and these were used in similar levels to those in MEM. DLS data showed that the nano Fe material remained mono-disperse in BSS with a hydrodynamic diameter in the low nanoscale (6.1 ± 0.3 nm, immediately following dispersion, PDI range 0.322–0.352) and that over a 24 h period the size of the material in suspension remained reasonably stable with an insignificant trend towards slight agglomeration/aggregation (figure 3(B)). Furthermore, the fractionation of the Fe into percentage of nanoparticulate, microparticulate and soluble Fe in the BSS suspension at pH 7.4 showed that almost all (≥95%) of the Fe was nanoparticulate, even after 24 h (figure 3(C)). There was no significant agglomeration/aggregation of the nanomaterial (i.e. formation of clusters of particles was <1%), and there was less than 4% soluble iron (figure 3(C)). The same was observed when BSS was used adjusted to pH 6.5, with only a small increase in the amount of soluble Fe observed for BSS adjusted to pH 5.8 (figure 3(D)).
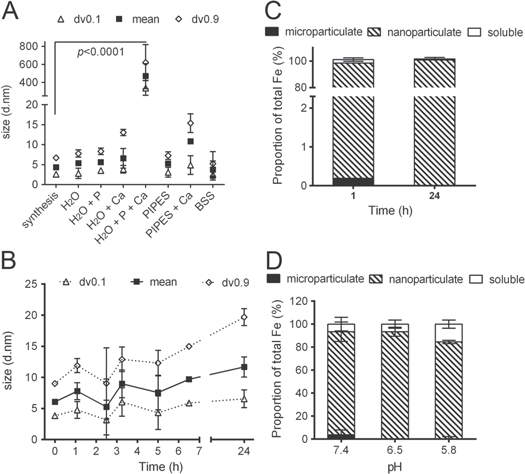
Figure 3. Particle size and phase distribution of nano Fe(III) oxo-hydroxide ('nano Fe') in different aqueous media. (A) Hydrodynamic diameter (DLS) of the nano Fe material 'as synthesized' or dispersed in different media at [Fe] = 1000 μM (this concentration was preferred here instead of 500 μM because the particle dispersions in H2O provided insufficient scattering). Synthesis=as synthesized; H2O= UHP water; P = sodium phosphate monobasic (1 mM); Ca = calcium chloride (1.8 mM); PIPES = 1,4-piperazinediethanesulfonic acid (10 mM); BSS = balanced salt solution containing 10 mM PIPES, 130 mM NaCl, 10 mM KCl, 1 mM MgSO4, 5 mM Glucose and 1.8 mM CaCl2. Data are reported in volume as mean (±SD) particle diameter, dv0.9 (±SD) and dv0.1 (±SD); n = 3 experimental replicates. p < 0.0001 for H2O+Ca+P compared to 'synthesis'. (B) Size stability over time of the nano Fe material dispersed in BSS ([Fe] = 500 μM). Data are shown as mean and size distribution (dv0.1 and dv0.9) of two independent replicates. Where not displayed the error bars are smaller than symbol size. Differences in mean diameter over time are not statistically significant (p ≥ 0.05). (C) Phase distribution of the material in BSS at 1 h and 24 h following initial dispersion ([Fe] = 500 μM). Data presented for the fractional percentage of microparticulate (black), nanoparticulate (hatched) and soluble Fe (white). Values are mean ±SD (n = 2 independent experiments). (D) Phase distribution of the material dispersed in BSS at different pHs ([Fe] = 500 μM). Data presented for the fractional percentage of microparticulate (black), nanoparticulate (hatched) and soluble Fe (white). Values are mean ±SD (n = 3 independent experiments). Detailed methodology is available in the methods section.
Download figure:
Standard image High-resolution imageFor more complex cellular uptake studies, where other components are supplemented into the medium, characterization of the nanomaterial in the new final (supplemented) environment is also necessary. Chemical agents that are used in cellular studies to inhibit or promote different uptake pathways of particles are commonly added to cell culture medium. Here we shown that some cause significant agglomeration or aggregation of the Fe(III) nanomaterial, whereas others showed little or no effect on the dispersion of the particles (figure 4). Mas-7, a stimulant of endocytosis [41]; cobalt, an inhibitor of iron uptake by the soluble Fe transporter DMT-1 [42, 43]; and chloroquine, which neutralizes lysosomal pH [44, 45], all led to almost complete agglomeration/aggregation of the nano Fe material in BSS at pH 7.4 (figure 4(A)). However, chloropromazine, an inhibitor of clathrin-mediated endocytosis [46, 47]; and monensin, an inhibitor of acidification of endosomes/lysosomes [48–50], did not significantly affect the nano-dispersion of the Fe oxide in BSS at pHs 7.4, 6.5 or 5.8 (figures 4(B) and (C))

{kind=link}
{kind=link}
{kind=link}
Figure 4. Phase distribution of nano Fe(III) oxo-hydroxide ('nano Fe') in BSS supplemented with several chemical inhibitors of cellular iron uptake. Throughout, data are presented for the fractional percentage of microparticulate (black), nanoparticulate (hatched) and soluble Fe (white) at [Fe] = 500 μM. (A) Phase distribution of the material dispersed in BSS at pH 7.4 supplemented with chloroquine, which neutralizes lysosomal pH [44, 45]; Mas-7 = mastoparan derivative, a stimulant of endocytosis [41]; Co(II) = cobalt, an inhibitor of iron uptake by the soluble Fe transporter DMT-1 [42, 43]; chloropromazine, an inhibitor of clathrin-mediated endocytosis [46, 47] or monensin, an inhibitor of acidification of endosomes/lysosomes [48–50]. Data from one experiment. (B) Phase distribution of the material dispersed in BSS at pH 5.8, 6.5 and 7.4 supplemented with 100 μM chloropromazine. Data are mean ±SD from three three independent experiments. (C) Phase distribution of the material dispersed in BSS at pH 5.8, 6.5 and 7.4 supplemented with 30 μM monensin. Data are mean ±SD from three three independent experiments. Detailed methodology is available in the methods section.
Download figure:
Standard image High-resolution image{kind=link}
We have used BSS for short-term iron uptake studies with nano Fe (up to 3 h exposure) in epithelial cell lines, namely Caco-2 and Hutu-80 cells, without any measured detrimental effect on cell viability or monolayer integrity as described elsewhere [20, 28, 51].
4. Conclusion
Here we provide the 'recipe' and basis of a simplified BSS that allows nano Fe to be added without marked agglomeration and/or aggregation over 24 h and is suitable for cellular assays involving incubation times with cells for at least up to 3 h.
The concept is applicable to other nanostructures such that even if the BSS developed here does not apply to all materials, the principles of how it was derived can be applied.
Acknowledgments
The authors would like to thank the UK Medical Research Council (MRC) (U105960399), Action Medical Research (SP4528) and WellChild for funding this work. BL thanks the MRC and Rank Prize Funds for her PhD studentship. This work is a publication of the UK Medical Research Council. We acknowledge Dr Andy Brown for collecting the TEM image and Dr Nuno Faria for his help with the DLS analysis.
The authors declare no conflict of interest.
Statement of authors' contributions to manuscript
DIAP, BL and JJP designed the research; DIAP and BL conducted the research and analysed data; DIAP had primary responsibility for final content. All authors read and approved the final manuscript.