
Abstract
We analysed the time evolution of several volatile organic compounds formed by the catabolism of ingested isotope-labelled ethanol using real-time breath gas analysis with proton-transfer-reaction mass spectrometry. Isotope labelling allowed distinguishing the emerging volatile metabolites from their naturally occurring, highly abundant counterparts in the human breath. Due to an extremely low detection limit of the employed technologies in the parts per trillion per volume range, it was possible to detect the emerging metabolic products in exhaled breath within ∼10 min after oral ingestion of isotope-labelled ethanol. We observed that ethanol was in part transformed into deuterated acetone and isoprene, reflecting the different fates of activated acetic acid (acetyl-coenzyme A), formed in ethanol metabolism. Using ethanol as a model clearly demonstrated the value of the here presented technique for the search for volatile markers for metabolic disorders in the exhaled breath and its potential usefulness in the diagnosis and monitoring of such diseases.
Export citation and abstract BibTeX RIS
1. Introduction
Breath gas analysis is a growing field of research with many potential and promising applications arising, as detailed in several review articles (Phillips 1992, Hermanns et al 1998, Schubert and Geiger 1999, Risby 2002, Miekisch et al 2004, Risby and Solga 2006, Buszewski et al 2007, Solga and Risby 2010, Amann et al 2011, Beauchamp 2011, Kwak and Preti 2011, Shirasu and Touhara 2011, Singer et al 2011, Spanel and Smith 2011). Exhaled human breath contains several hundreds of volatile organic compounds (VOCs), (Pauling et al 1971, Phillips et al 1999, Kushch et al 2008b, Kohl et al 2013) which might serve—alone or in combination-–as biomarkers in medicine.
To date the knowledge on the origin and fate of most of the breath VOCs is rare. The mechanisms of the formation and metabolism of VOCs are not understood in detail so far, even for extensively studied compounds such as isoprene (Kushch et al 2008a, King et al 2009, 2010, 2012, Fuchs et al 2012).
In recent years, several breath tests have emerged to detect infections, metabolic disorders and food intolerances, with the help of ingested isotope-labelled substances (Kurpad et al 2002, Modak 2009). Many of these breath tests are based on the detection of the metabolically produced 13C-isotope of CO2.
Here, we present the results of the assessment of a procedure to monitor a multitude of intermediate metabolic products after the ingestion of isotope-labelled substances. We utilized the well-understood metabolic degradation of ethanol as a study model. Highly sensitive proton-transfer-reaction mass spectrometry (PTR-MS) served for real-time monitoring of the emerging metabolic volatile products simultaneously. Due to its very low detection limit of less than 50 parts per trillion per volume (pptv), the different metabolic VOCs could be detected immediately at their onset of production, or at early times in the course of metabolism, respectively. High-resolution PTR time-of-flight mass spectrometry (PTR-TOF-MS) was used for the determination of the exact ion masses of the protonated molecules to enable the identification of the metabolites.
Results from previous breath gas ethanol catabolism studies (Spanel et al 2005) suggest that in order to observe metabolites that stem from the oxidation of previously ingested ethanol, a specific approach is needed. Using isotopically labelled ethanol allowed us to distinguish the emerging labelled VOCs from their naturally occurring counterparts in the exhaled human breath. This is especially important when searching for small variations in such substances, which are always present at rather high and varying concentrations in exhaled breath gas, such as acetone and isoprene.
The reported experiments reveal that ethanol was, in part, metabolized to acetone and isoprene, as deuterated acetone and isoprene were observed. This reflects the different metabolic paths for activated acetic acid (acetyl-coenzyme A), an important ethanol metabolite.
Our results show the advantage of monitoring the intermediate metabolites. Instead of waiting for the oxidative end-product CO2 to emerge, much shorter times are involved in detecting the intermediate metabolic products.
2. Materials and methods
2.1. Sensors for breath gas analysis
The chemical ionization technique of PTR-MS (Ionicon Analytik, Innsbruck, Austria) was used for breath gas analysis in this study. Due to its short measurement response time of less than 100 ms and its high sensitivity for a large number of VOCs, PTR-MS was ideal for real-time monitoring of evolving compounds. PTR-MS is an already well-established analytical method in many fields of VOC analysis at trace gas concentrations and has been described extensively elsewhere (de Gouw et al 2003, Schwarz et al 2009, Fierens et al 2012). The technique produces a distinctive product ion on a mass-to-charge (m/z) ratio of 1 amu higher than the molecular mass of the neutral compound. As an example, ethanol (molecular mass = 46 u) produces a signal on m/z 47 in the mass spectrum. Due to its soft ionization technique, PTR-MS offers rather simple spectra, even if a large number of different VOCs are present in a sample.
In addition, a PTR-TOF-MS (Jordan et al 2009, Graus et al 2010, Kohl et al 2013) (Ionicon Analytik, Innsbruck, Austria) was used to determine the molecular masses of the observed metabolites with high accuracy. In PTR-TOF-MS, the mass resolution (at full width at half maximum) m/Δm is greater than 4000, which is sufficient to resolve the protonated molecules formed from ethanol (m/z 47.0497, C2H7O+) and formic acid (m/z 47.0133, CH3O2+).
2.2. Breath gas sampling
An important prerequisite for the accurate determination of the concentrations of breath gas trace compounds is the correct sampling of the exhaled breath gas. For this purpose, we employed a buffered end-tidal (BET) breath gas sampler (Ionimed Analytik, Innsbruck, Austria) which was described in detail by Herbig et al (2008). In short, the BET breath gas sampler consists of a so-called buffering tube, a tube made of perfluoroalkoxy co-polymer of ∼30 cm length and 1 cm inner diameter, and a capillary that connects the PTR-MS instrument to the centre of the buffering tube. One side of the buffering tube carries the mouthpiece, while the other side is open to the environment. In order to eliminate water condensation and minimize surface affinity of certain chemical compounds, the entire BET breath gas sampler was heated to 80 °C while operation. For safety reasons, the mouthpiece remained unheated.
2.3. Procedures
Three healthy volunteers participated in the search for ethanol metabolites in the breath. They gave written informed consent to participate in this non-invasive study. The test subjects were all white male Europeans with a body mass index between 22 and 26 and were between 33 and 36 years of age. The test subjects did not eat breakfast before the tests started at around 9 a.m. Lunch and dinner were allowed at usual times.
The test subjects were asked to fully exhale into the buffering tube of the BET breath gas sampler and then to withdraw from the mouthpiece before inhaling. After providing a complete exhalation, the test subject sat next to the BET while breathing normally. In this way, the last fraction of the exhaled breath gas stayed inside the buffering tube and was gradually transported towards the low-pressure region of the ionization chamber of the PTR-MS instrument, which was continuously monitoring and recording data. Gradually, the exhaled breath gas sample was replaced by room air in the buffering tube. After the PTR-MS signals had returned to room air levels, the test subject provided another complete exhalation into the BET. This protocol resulted in the measurement of two exhalations per minute.
PTR-MS continuously measured 10–15 different ions (associated with ethanol, acetaldehyde, acetone and isoprene) per measurement cycle, at an integration time of 50–200 ms per ion. We additionally monitored the humidity in every cycle by measuring the protonated water-dimer (H3O+ ⋅ H2O) signal at m/z 37 (with a detection time of 50 ms). The hydronium primary ion signal (H3O+, measured via the 18O isotope on m/z 21) was always determined (with 50 ms integration time) for signal normalization purposes. The protonated water-dimer signal varied greatly within a breathing cycle due to large differences in the humidity between exhaled breath gas and ambient air. We used the water-dimer signal to identify the end tidal fraction of each exhalation, which is unambiguous, because the absolute humidity of exhaled breath is known to be ∼6% at normal body temperature (Spanel and Smith 2001), which is much higher than the humidity in ambient air. With a simple mathematical algorithm, the end-tidal part was determined for each exhalation and the corresponding mean values and standard deviations were calculated for the identified time window.
2.4. Calibration
The PTR-MS was calibrated using a commercially available gas calibration system (GCU-a, Ionimed Analytik, Innsbruck, Austria) (Singer et al 2007). This system provided a pure gas stream with an adjustable quantity of calibration gas (0–100 ppbv) and variable humidity. The calibration gas used contained the relevant VOCs in this study, which included acetaldehyde, ethanol, acetone and isoprene. The sensitivity factors for the PTR-MS were 16.0 ncps/ppbv (normalized counts per second/parts per billion in volume, count rate normalized with respect to the precursor ions) for acetaldehyde (m/z 45), 0.4 ncps/ppbv for ethanol (m/z 47), 22.0 ncps/ppbv for acetone (m/z 59) and 4.8 ncps/ppbv for isoprene (m/z 69). The calibrated sensitivity factors were used to calculate the reported mixing ratios.
2.5. Isotope-labelled ethanol
D3-ethanol (CD3–CH2–OH) (DeuChem, 99.0%) was used. The D3-ethanol was filled in medical gelatine capsules and ingested together with some water. The gelatine quickly dissolved in the stomach and released the ethanol. This procedure avoided ethanol contamination of the mouth space. The typically ingested D3-ethanol dose was 6 µl per 1 kg body mass, i.e. 450 µl for a person with a mass of 75 kg. This is ∼1/25 of the alcohol quantity that one ingests when drinking a small glass of wine.
2.6. Mathematical modelling of the change in signal with time
Simple mathematical models were used to describe the time evolution of the measured VOC signals.
Concentration variations for several compounds were found to be well described by a simple exponential
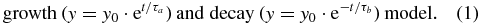
In the cases where the exponential function did not suitably reproduce the mass signal's variation in time, we used a different model that implied that the measured metabolic product P was produced from a precursor A with a rate ka and was removed with a rate kp:
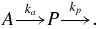
Such a system can be described by the following differential equation system:
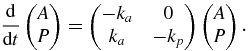
The solution for P can be found with the method called 'variation of the constant' (Atkins and de Paula 2009) and is
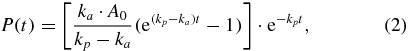
with A0 being the concentration of the precursor A at time t = 0.
This simple model showed a good agreement with the observed signals and was sufficient to describe their time constants (τa = 1/ka, τp = 1/kp).
For some signals, an extended model, assuming a compound function consisting of two parts, had to be used:
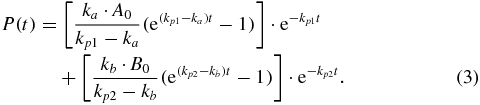
3. Results
3.1. Ethanol
Initial experiments revealed that small amounts of ingested D3-ethanol did not lead to an elevated ethanol signal (at m/z 50) in exhaled breath. Such small amounts of ethanol are highly likely catabolized by gastric enzyme alcohol dehydrogenase (ADH) (Lieber 1997) before reaching the circulating blood. The following procedure was used for the experiments: the test subjects first drank up to 10 ml of normal, unlabelled ethanol (Sigma Aldrich, 99.5% purity), leading to an increase in exhaled ethanol signal at m/z 47. Only then the test subjects ingested a capsule containing a small amount of labelled ethanol. Using this protocol, the labelled and unlabelled ethanol breath signals showed very similar variations in time, using PTR-MS (figure 1). The increase and the decrease of both occurred on a timescale τ of about 20 min, as determined by model (1). From this, we infer that the uptake and removal mechanisms are the same for unlabelled and labelled ethanol.

Figure 1. PTR-MS signals at m/z 47 (red circles) and m/z 50 (blue squares) resulting from breath gas analysis after the ingestion of 10 ml unlabelled ethanol (orally ingested solution diluted in water) and 500 µl D3-ethanol (ingested capsule). The vertical black line marks the time of ingestion of the unlabelled ethanol, and the vertical green dashed line marks the time of ingestion of the D3-ethanol.
Download figure:
Standard image High-resolution imageThe maximum concentration of the unlabelled ethanol signal at m/z 47 was ∼35 ppmv, which corresponds to a blood alcohol level of 0.13‰ if we assume an ethanol blood/breath ratio of 2000/1 and the calculations described by Jones et al (1992) and Hlastala (1998). This result is consistent with the expected blood alcohol concentration calculated by the Widmark formula (Widmark 1932): c = A/(p ⋅ r) ≈ 0.13‰, with A being the mass of the ingested ethanol (A = 7.9 g), p the test person's body mass (p = 80 kg) and a reduction factor r = 0.76 of the test subject (Watson et al 1980).
The maximum concentration of the labelled ethanol PTR-MS signal at m/z 50 was ∼160 ppbv, corresponding to a blood alcohol level of 0.0006‰, which is ∼10 times less than that calculated by the Widmark formula. This may be explained by a loss of the D-3 ethanol due to H/D exchange with water or other molecules. In fact, we observed PTR-MS signals at m/z 48 and 49, which exhibit a similar temporal profile to that of m/z 50. This result suggests that m/z 49 and m/z 48 may reflect the presence of D2- and D1-ethanol. The maximum concentration of the D2-isomer in exhaled breath was approximately the same as the D3-isomer's concentration and the D1-ethanol was approx. doubled in concentration compared to the D3-isomer. Complete H/D exchange leading to the formation of 'normal' ethanol could have made up for the missing fraction, which would be indistinguishable from ethanol from other sources.
3.2. m/z 61 to 66
The PTR-MS signals at m/z 62, 63, 64 and 65 changed in time after D3-ethanol ingestion, as seen by real-time monitoring with PTR-MS. Using PTR-TOF-MS, the exact masses of these compounds were determined to be m/z 62.068, 63.074, 64.081 and 65.087 (±0.002). Within the instruments accuracy (Kohl et al 2013), this was consistent with the calculated m/z of protonated isotopologues of C3DxHyO ⋅ H+ with x + y = 6, namely m/z 62.0685 (C3H3D3O ⋅ H+), 63.074 (C3H2D4O ⋅ H+), 64.0811 (C3HD5O ⋅ H+) and 65.0874 (C3D6O ⋅ H+).
These signals were most probably caused by isotopologues of acetone, but we want to remind the reader that different D-isotopologues of propanal (C3H6O), propenol (C3H6O) and propanol (C3H8O) are also possible. Acetone, however, is the biochemically most logical compound.
Analysis of the variation in time (see figure 2) of the PTR-MS data revealed differences in the kinetic of formation and removal between these isotopologues. In particular, m/z 63 and 64 behaved very differently from m/z 62 and 65.

Figure 2. PTR-MS data of m/z 62 (a), 64 (b) and 65 (c) resulting from breath gas measurement after the ingestion of a gelatine capsule containing 500 µl D3-ethanol. The red dots represent the end-tidal breath gas values of the test subject, the blue dots the inhale values, i.e. the room air. The black line is the fit of model (2) to the data points. For m/z 62 and m/z 65 the extended model (3) had to be used. The green vertical line marks the time of ingestion of the D3-ethanol.
Download figure:
Standard image High-resolution imageThe signal at m/z 62 (C3H3D3O ⋅ H+) (figure 2(a)) showed a very fast increase (τa ≈ 15 min) and decreased with an initially fast (τp1 ≈ 1.5 h) and then slow rate (τp2 ≈ 25 h). The signal at m/z 63 (C3H2D4O ⋅ H+) (not shown) exhibited a similar behaviour like m/z 64. The signal at m/z 64 (C3HD5O ⋅ H+) (figure 2(b)) increased over several hours (τa ≈ 6 h) and decreased with a slow rate (τp ≈ 10 h). The strongest signal was at m/z 65 (C3D6O ⋅ H+) (figure 2(c)) which showed a very short rise time (τa ≈ 15 min) similar to m/z 62. The removal of m/z 65 followed a fast (τp1 ≈ 15 min) and a long timescale of several hours (τp2 ≈ 8 h). We note that it is possible that different timescales of the decreases were caused by an overlap of two occurrences from two different pathways.
The signal of the m/z 65' natural 13C-isotope with an abundance of 3.3% was found at m/z 66 and showed an identical variation in time. It corresponded to a concentration far below 1 ppbv (see figure 3). The limit of detection (LOD)1 for m/z 66 was about 50 pptv with an integration time of 200 ms.

Figure 3. PTR-MS data of m/z 65 (a) and m/z 66 (b) of a breath gas measurement after ingestion of 500 µl D3-ethanol. The red dots are the breath gas values of the test subject, the blue dots the concentrations in ambient air (=inhaled concentrations). The black line is a fit of model (3) to the data points. The vertical green dashed line marks the point in time of the D3-ethanol ingestion. M/z 66 is the natural isotope (with 3.3% abundance) of m/z 65. This signal at m/z = 66 demonstrates clearly the possibility of measuring metabolites in the breath with concentrations far below 1 ppbv (LOD ≈ 50 pptv).
Download figure:
Standard image High-resolution imageWe observed a small signal at m/z 61.056 (not shown) using PTR-TOF-MS, that first increased and then decreased after isotope-labelled ethanol ingestion. The measured mass does not allow an unambiguous attribution, but either acetone (C3H4D2O ⋅ H+) or propenal (C3D4O ⋅ H+) are possible. The acetic acid signal (m/z 61.029) is well separated from m/z 61.056.
3.3. Isoprene
We detected signals at m/z 70, 71 and 72 using PTR-MS, with all of them showing similar variations in time after ethanol ingestion (see figure 4). With PTR-TOF-MS, the exact masses were determined to be m/z 70.075, 71.084 and 72.089 (±0.002). This was consistent with the calculated masses of the protonated isotopologues of C5H8 with one, two or three deuterium atoms namely m/z 70.0767 (C5H7D1 ⋅ H+), 71.0830 (C5H6D2 ⋅ H+) and 72.0893 (C5H5D3 ⋅ H+). These compounds were most probably isotope-labelled isoprene.

Figure 4. PTR-MS data of m/z 70, 71 and 72 of a breath gas measurement after ingestion of D3-ethanol. The green vertical line marks the time of ingestion.
Download figure:
Standard image High-resolution imageM/z 70 had a similar signal strength as m/z 71. M/z 72 was about 15% of the signal intensity of m/z 71. The onset of a signal increase could be detected after ∼20 min after the D3-ethanol ingestion. The signals increased with a timescale τa of about 0.5 h and decrease on a timescale τb of about 1 h. The time constants τa and τb for m/z 70, 71 and 72 were determined by model (1).
3.4. Inter-individual differences
Figure 5 shows the time variations of the PTR-MS signals at m/z 62, 64, 65 and 70 of three different individuals. The signals differed mainly with respect to their intensity. The maximum concentration of m/z 62 ranged from 1.4 to 4.2 ppbv, for m/z 64 it ranged from 1.5 to 3 ppbv, for m/z 65 it ranged from 16 to 26 ppbv and for m/z 70 it ranged from 4 to 16 ppbv. However, the signals' variation in time (characterized by τ) was very similar and the maxima coincided in time too, pointing to the fact that the kinetic of formation and removal of the underlying compounds were the same in different individuals.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. PTR-MS data of m/z 62 (a), 64 (b), 65 (c) and 70 (d) of three different individuals (P1, P2, P3) after D3-ethanol ingestion (6 µl per 1 kg body mass). The coloured dots represent the end-tidal breath gas values of the different test subjects. The coloured solid lines are fits of model (2) and (3) to the corresponding data points (not used for m/z 70 in (d)). The vertical green dashed line marks the point in time of the D3-ethanol ingestion.
Download figure:
Standard image High-resolution image{kind=link}
4. Discussion and conclusion
We observed the time variations of MS signals of VOCs after ethanol ingestion in the exhaled breath gas of three healthy individuals. In order to recognize metabolites that stem from the ingested ethanol, we used deuterium-labelled ethanol. The test subjects exhibit normal drinking behaviour; therefore, ethanol is expected to be mainly eliminated from the body by oxidative degradation in the liver cells (Strohle et al 2012). Step 1 is the oxidation to toxic acetaldehyde mediated by the ADH. Next, acetaldehyde is transformed into acetic acid catalyzed by the enzyme aldehyde dehydrogenase. Finally, acetate is activated via a chemical bond to form acetyl-coenzyme A.
There were no differences in the kinetic of the uptake/formation and removal of the observed signals between the tested individuals, supporting that—despite differences in absolute concentrations of these VOCs—the same metabolic pathways were used to rid the body from ethanol and its metabolites.
We did not observe a signal of the deuterium-labelled acetaldehyde. We conjecture that this toxic and highly reactive intermediate product did not enter the blood stream but was quickly further metabolized. Ingested D3-ethanol was in part converted into acetone (or other possible isomers and isobars) and isoprene, as proven by the increase of the corresponding signals (figures 2 and 3) in the mass spectra. The formation of these metabolites in the body is accomplished via acetyl-CoA, which is formed in ethanol oxidation.
Acetyl-CoA is further used in different biochemical strategies. It is either introduced into the citric acid cycle to produce CO2, H2O and energy, but it is also used in the synthesis of bio-molecules, such as fatty acids. On the other side, acetone is formed in fat catabolism in the liver, where acetyl-CoA is transformed into so-called ketone bodies, like acetoacetate, which is further reduced to 3-hydroxybutyrate or slowly decarboxylizes to acetone. Acetoacetate is an important energy source in times of starvation or in people suffering from diabetes. Isoprene is known to be produced in the mevalonic pathway, and is an intermediate product in the biosyntheses of e.g. cholesterol from acetyl-CoA (Berg et al 2007).
We found two very different kinetic behaviours for the detected signals at m/z 62, 63, 64 and 65 (see figure 5): m/z 62 and m/z 65 exhibit a very fast kinetic in formation and removal and m/z 63 and 64 exhibit a slow kinetic of formation and removal. From the exact masses, derived by PTR-TOF-MS, we know that either deuterium-labelled acetone or its isomers (propenol and propanal) and isobars (propanol) are possible compounds. The acetoacetate/acetone formation requires in total three acetyl-CoA, and combinations of acetic acid containing different numbers of deuterium atoms are imaginable, leading to the formation of D3- to D6-acetone. However, we cannot explain right away the finding of such different timescales in formation and removal. It is feasible that two different metabolic pathways lead to the formation of acetone with either four and five deuterium atoms (slow kinetic) or three and six deuterium atoms (fast kinetic). Deuterium loss in the precursors in the case of D4 and D5 might cause longer times of formation, but this would not explain the slow removal. It is also possible that the product of the fast formation and removal kinetic represents a different chemical compound than the product of the slow kinetic.
In order to connect signals to specific metabolic paths, it was necessary to identify the underlying compounds. High-mass-resolving PTR-TOF-MS was used to determine the observed metabolite's masses with high accuracy, allowing us to derive possible atomic compositions. For the m/z range between 61 and 66, we could not unambiguously identify a specific compound. Several chemical compounds of similar or identical masses, such as propenol, propanal, acetone and propanol come into question. From a biochemical perspective, acetone makes most sense.
The formation and removal of a volatile metabolite was characterized by applying different fit models to the measured curves. As a result, τ was characteristic for the same m/z in different individuals, independent from the absolute concentrations of the respective compounds. This in agreement with the work of Halbritter et al (2012), who performed breath gas analyses with pregnant woman during an oral glucose tolerance test. The gestation diabetes diagnosis could be mapped to the characteristic time evolution of the volatile oxidation products of glucose and lipids, acetone metabolites and thiols after a glucose challenge.
The attempt of a challenge test is to distinguish volatile compounds formed in response to a certain challenge from volatile compounds that stem from other sources. The testing time of such a challenge test should be short enough and the setting should be in such a way to keep all other breath gas components constant. That way, the latter can be treated as a constant background even if the observed metabolite lies at the same m/z like another VOC that will not be changed by the challenge.
Our results clearly demonstrate the capability of PTR-MS to follow several metabolites throughout their formation and metabolic removal in real time. Arising metabolites could be detected at an early stage of their formation due to PTR-MS's high sensitivity. It therefore bears the potential to detect and monitor metabolic diseases by analysing a cascade of formed metabolites along particular biochemical pathways.
Acknowledgments
The research leading to these results has received funding from the FFG (Forschungsförderungsgesellschaft mbH, Austria). The authors thank the volunteers for their participation in this study.
Footnotes
- 1
LOD = 3 ⋅ σ.