Abstract
Aspartic acid (Asp) residues in peptides and proteins (l-Asp) are known to undergo spontaneous nonenzymatic reactions to form l-β-Asp, d-Asp, and d-β-Asp residues. The formation of these abnormal Asp residues in proteins may affect their three-dimensional structures and hence their properties and functions. Indeed, the reactions have been thought to contribute to aging and pathologies. Most of the above reactions of the l-Asp residues proceed via a cyclic succinimide intermediate. In this paper, a novel three-water-assisted mechanism is proposed for cyclization of an Asp residue (forming a gem-diol precursor of the succinimide) by the B3LYP/6-31 + G(d,p) density functional theory calculations carried out for an Asp-containing model compound (Ace−Asp−Nme, where Ace = acetyl and Nme = NHCH3). The three water molecules act as catalysts by mediating 'long-range' proton transfers. In the proposed mechanism, the amide group on the C-terminal side of the Asp residue is first converted to the tautomeric iminol form (iminolization). Then, reorientation of a water molecule and a conformational change occur successively, followed by the nucleophilic attack of the iminol nitrogen on the carboxyl carbon of the Asp side chain to form the gem-diol species. A satisfactory agreement was obtained between the calculated and experimental energetics.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Introduction
Aspartic acid (Asp or D) residues in peptides and proteins (l-Asp) are known to undergo spontaneous nonenzymatic reactions to form l-β-Asp, d-Asp and d-β-Asp residues via a cyclic succinimide intermediate as shown in figure 1 (Geiger and Clarke 1987, Stephenson and Clarke 1989, Fujii et al 1999, Cloos et al 2003, Fujii and Saito 2004, McCudden and Kraus 2006). The formation of these abnormal amino acid residues in proteins may affect their three-dimensional structures and hence their properties and functions. Thus, the reactions have been thought to play important roles in aging and pathologies, especially those of age-related diseases such as cataracts and Alzheimer's disease (Ritz-Timme and Collins 2002, Fujii 2005, Hipkiss 2006, Truscott 2010, Hooi and Truscott 2011).
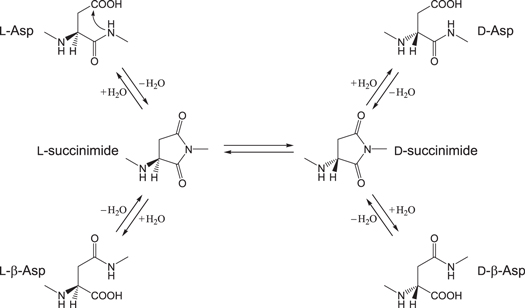
Figure 1. Succinimide-mediated reactions of an Asp residue.
Download figure:
Standard image High-resolution imageThe rate-pH profile for the Asp to β-Asp isomerization of the hexapeptide YADGFK shows no remarkable pH dependence (Yi et al 2013). In particular, the isomerization rate is almost pH-independent in the range of pH 6–8, which indicates that the reaction is uncatalyzed or, much more probably, water-catalyzed. Indeed, the kinetic data shows that, at the physiological pH of 7.4, more than 99% of the total isomerization is uncatalyzed or water-catalyzed.
A density functional theory (DFT) study has recently been reported for uncatalyzed and one-water-catalyzed two-step mechanisms for the succinimide formation from an Asp residue (Catak et al 2009). The first step is cyclization (intramolecular nucleophilic attack by the C-terminal amide nitrogen on the side-chain carboxyl carbon) to form a gem-diol tetrahedral intermediate, and the second step is dehydration from the intermediate. It was shown that the first step is rate-determining in both the mechanisms, and that the single water molecule largely decreases the activation barrier. However, the calculated free energy of activation for the one-water-catalyzed mechanism was 46.1 kcal mol−1, which is still too high for a reaction which occurs under the physiological temperature of 37 °C on biologically relevant time scales (up to decades). Indeed, from the Arrhenius plot for the succinimide formation in a human αA-crystalline peptide TVLDSGISEVR under physiological conditions (Aki et al 2013), the activation energy for this reaction can be calculated to be 25.6 kcal mol−1. Another experimental activation energy estimated for the succinimide formation in the hexapeptide VYPDGA is even lower (21.7 kcal mol−1) (Geiger and Clarke 1987).
Normally, amide nitrogens are poor nucleophiles due to the resonance shown in figure 2. We have recently shown by DFT calculations that the isomerization of the C-terminal amide group of an Asp residue to the tautomeric iminol form (iminolization) can readily occur with assistance of two water molecules and the protonated carboxyl group of the Asp side chain (figure 3), and that cyclization from the iminol tautomer (which is also catalyzed by two water molecules) is favored over the direct cyclization from the amide form (Takahashi and Oda 2012). The amide−iminol tautomerism (figure 2) is a nitrogen analogue of the well-known keto−enol tautomerism. It is expected from the resonance of the iminol group shown in figure 2 that an iminol nitrogen is much more nucleophilic than the corresponding amide nitrogen. Thus, we have proposed that the cyclization to form the gem-diol precursor of the succinimide intermediate occurs after the C-terminal amide group is converted to the iminol form (figure 4). This means that the long-established mechanism of the succinimide formation from Asp residues (cyclization−dehydration) may have to be modified by including the iminolization step (iminolization−cyclization−dehydration).
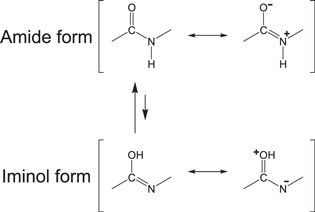
Figure 2. Amide−iminol tautomerism and resonance structures for the amide form (upper) and the iminol form (lower).
Download figure:
Standard image High-resolution image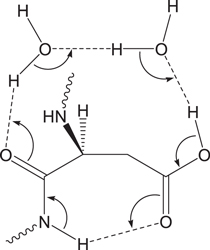
Figure 3. Bond reorganization in the iminolization of the C-terminal amide group of an Asp residue assisted by two water molecules and the side-chain carboxyl group (protonated form).
Download figure:
Standard image High-resolution image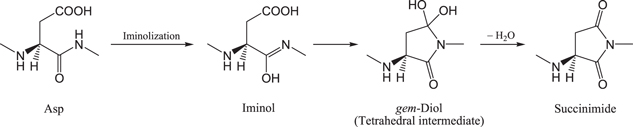
Figure 4. The iminolization−cyclization−dehydration mechanism of the succinimide formation from an Asp residue.
Download figure:
Standard image High-resolution imageIn this paper, we propose, based on DFT calculations, another water-assisted mechanism for the formation of the gem-diol species from Asp residues, where three water molecules catalyze the iminolization and cyclization steps by enabling 'long-range' proton transfers. The three-water-catalyzed mechanism for the amide−iminol tautomerism has recently been proposed based on DFT and ab initio calculations for N-methylacetamide as a model compound (Li et al 2008). In this mechanism, the three water molecules form a hydrogen-bond bridge between the oxygen and NH hydrogen atoms of the amide group (figure 5), along which a quadruple proton transfer occurs, i.e., four protons are transferred, concerted with the O=C−N to O−C=N bond reorganization.
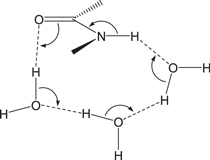
Figure 5. Bond reorganization in the iminolization of an amide group assisted by three water molecules.
Download figure:
Standard image High-resolution imageMethods
All calculations were performed using Spartan'08 (Wavefunction, Irvine, CA). Figure 6 shows the model compound used in the present study, in which an Asp residue is capped with an acetyl (Ace) group and an NHCH3 (Nme) group on the C- and N-termini, respectively. This compound has been used in previous computational studies (Catak et al 2006, 2009, Takahashi and Oda 2012) related to the present one. Only the protonated Asp residues are thought to undergo the succinimide-forming reaction, because the deprotonated carboxyl group has a reduced electrophilicity and no suitable leaving group (Stephenson and Clarke 1989, Capasso 1996, Takahashi and Oda 2012, Yi et al 2013). Therefore, the side-chain carboxyl group was taken as protonated. The ϕ (C−N−Cα −C) and ψ (N−Cα −C−N) dihedral angles characterize the main-chain conformation, and the χ1 dihedral angle (N−Cα −Cβ −Cγ ) characterizes the side-chain conformation. Three water molecules were placed to form the reactant complex in which they form a hydrogen-bond bridge between the oxygen and NH hydrogen atoms of the amide group on the C-terminal side of the Asp residue.
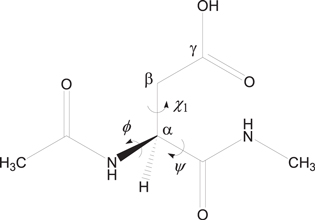
Figure 6. The model compound (Ace−Asp−Nme) used in the psesent study. The ϕ (C−N−Cα −C) and ψ (N−Cα −C−N) dihedral angles which characterize the main-chain conformation and the χ1 dihedral angle (N−Cα −Cβ −Cγ ) which characterizes the side-chain conformation are indicated.
Download figure:
Standard image High-resolution imageEnergy-minimum and transition state (TS) geometries were located without any constraints by the DFT calculation with the B3LYP functional and the 6-31 + G(d,p) basis set. This method has been successfully used for related reaction systems (Catak et al 2006, 2009, Takahashi et al 2010, Takahashi and Oda 2012, Takahashi 2013). It was confirmed by vibrational frequency calculations that there is no imaginary frequency for the energy-minimum geometries and there is a single imaginary frequency at each TS. Also, the relative energies were corrected for the zero-point energy (ZPE).
Results and discussion
A four-step reaction pathway was found starting from the reactant complex AM (amide form). Figure 7 shows the energy diagram for the entire reaction. The imaginary frequencies (cm−1) of the TSs are also shown in this figure. IM1, IM2 and IM3 are different complexes between the iminol form and the three water molecules. TSAI is the TS connecting AM and IM1. The TS between IM1 and IM2 is denoted as TSIM12, and that between IM2 and IM3 as TSIM23. TH stands for the gem-diol tetrahedral species (complex with three water molecules), and TSIT is the TS connecting IM3 and TH. The optimized geometries are shown in figures 8–16. Geometrical parameters relevant to the following discussions are listed in table 1.

Figure 7. Energy diagram for the three-water-assisted formation of the gem-diol tetrahedral species (TH) from the reactant complex (AM) via the iminol form (IM1, IM2 and IM3). The ZPE-corrected relative energies (kcal mol−1) with respect to AM are shown for the optimized geometries (the total energy of AM is −913.825 849 Eh). The single imaginary frequency (cm−1) is also shown for each TS geometry.
Download figure:
Standard image High-resolution image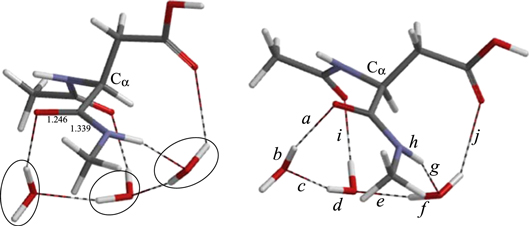
Figure 8. Two views of the optimized geometry of the reactant complex AM. The three catalytic water molecules are circled in the left figure. Bond lengths shown are in Å. For the values of the interatomic distances a−j, see table 1.
Download figure:
Standard image High-resolution imageTable 1. The ϕ, ψ and χ1 dihedral angles (°) and selected interatomic distances (Å) of the optimized geometries. For the definitions of the distances a−k, see figures 8– 16.
| Geometry | ϕ | ψ | χ1 | a | b | c | d | e | f | g | h | i | j | k |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AM | −120 | 158 | −158 | 2.001 | 0.976 | 1.995 | 0.975 | 1.821 | 0.985 | 1.817 | 1.032 | 1.976 | 2.387 | 2.797 |
| TSAI | −90 | 180 | −155 | 1.344 | 1.103 | 1.217 | 1.203 | 1.012 | 1.625 | 1.001 | 1.776 | 1.712 | 2.413 | 2.736 |
| IM1 | −86 | 180 | −156 | 1.001 | 1.661 | 1.000 | 1.682 | 0.982 | 1.856 | 0.981 | 1.941 | 1.965 | 2.498 | 2.709 |
| TSIM12 | −86 | −166 | −161 | 0.999 | 1.663 | 0.998 | 1.692 | 0.980 | 1.850 | 0.973 | 2.554 | 1.965 | 2.974 | 2.176 |
| IM2 | −95 | −147 | −165 | 0.999 | 1.658 | 0.998 | 1.690 | 0.980 | 1.826 | 0.976 | 3.762 | 1.968 | 3.242 | 1.870 |
| TSIM23 | −106 | −140 | −168 | 1.006 | 1.611 | 1.001 | 1.666 | 0.980 | 1.821 | 0.977 | 4.156 | 1.925 | 3.251 | 1.856 |
| IM3 | −148 | −115 | −173 | 1.001 | 1.655 | 1.001 | 1.657 | 0.981 | 1.810 | 0.977 | 4.552 | 1.936 | 3.246 | 1.858 |
| TSIT | −121 | −141 | 158 | 1.273 | 1.144 | 1.127 | 1.306 | 1.009 | 1.615 | 1.034 | 2.985 | 1.827 | 2.988 | 1.525 |
| TH | −128 | −144 | 151 | 1.804 | 0.983 | 1.827 | 0.982 | 1.684 | 0.997 | 1.727 | 2.593 | 1.958 | 3.255 | 0.991 |
Iminolization
The geometry of the reactant comlex AM is shown in figure 8. The ϕ, ψ and χ1 dihedral angles (see figure 6) are −120°, 158° and −158°, respectively. A chain of the three water molecules forms a hydrogen-bond bridge between the oxygen and NH hydrogen atoms of the C-terminal amide group. Thus, a cyclic structure is formed by the amide group and the three water molecules. The middle water molecule also form a hydrogen bond (1.976 Å) to the oxygen of the N-terminal amide group (bond i in figure 8). There is also a weak hydrogen bond (2.387 Å) between the water molecule hydrogen-bonded to the amide NH and the carbonyl oxygen of the side chain (bond j in figure 8).
The first step in figure 7 is iminolization of the C-terminal amide group to form IM1 (figure 10), which is 16.2 kcal mol−1 higher in energy than AM. The three water molecules enable a quadruple proton transfer by which the conversion of the amide group to the iminol form occurs (figure 5). As mentioned in the Introduction, the nucleophilicity of the nitrogen atom is expected to be greatly reinforced by the iminoliztion. The calculated activation barrier of the iminolization is 21.2 kcal mol−1, which is possible for a biological reaction. In the corresponding TS, denoted as TSAI (figure 9), the hydrogen bond to the N-terminal amide oxygen (bond i in figure 9) is considerably shorter (1.712 Å) than in AM, indicating that this hydrogen bond contributes to stabilization of the TS. The quadruple proton transfer is asynchronous. From the interatomic distances a−h in TSAI (see table 1 and figure 9), it may be said that elongation of the amide NH bond leads the entire proton transfer. In TSAI, the proton originally located in the NH bond has moved near to its new position. It is notable that, in IM1, the hydrogen bond between the iminol OH group and one of the water molecules (bond b in figure 10) is significantly short (1.661 Å) (and the neighboring OH bonds, a and c, are long). On going from AM to TSAI, ϕ and ψ increase by 30° and 22°, respectively, while χ1 increases by only 3° (table 1). These dihedral angles are very similar between TSAI and IM1.

Figure 9. The geometry of TSAI. Bond lengths shown are in Å.
Download figure:
Standard image High-resolution image
Figure 10. Two views of the geometry of IM1. Bond lengths shown are in Å.
Download figure:
Standard image High-resolution imageIn the iminol moiety of IM1, the OH group is 'cis' with respect to the C=N bond (the NCOH dihedral angle is −45°). This large deviation from planarity is due to the closeness of the nearby methyl group. The distance between the iminol hydrogen and one of the hydrogens of the methyl group is as short as 2.345 Å in IM1.
Hydrogen bond rearrangement and conformational change in the iminol form
In IM1, the distance between the iminol nitrogen and the carboxyl carbon of the side chain is 3.046 Å. This value is shorter than the sum (3.25 Å) of the van der Waals radii of the two atoms (C = 1.70 Å, N = 1.55 Å) (Bondi 1964). However, we could not found a cyclization TS which directly connected to IM1. This is probably in part due to the hydrogen bond (1.941 Å) between the iminol nitrogen and one of the water molecule (bond h in figure 10). This hinders the motion required to make a covalent bond between the nitrogen and carbon atoms and also reduces the nucleophilicity of the nitrogen atom. This hydrogen bond is broken by reorientation of the water molecule to give IM2 (figure 12) which occurs via TSIM12 (figure 11). In IM1, this water molecule had also a weak hydrogen bond (2.498 Å) to the carboxyl carbon of the side chain (bond j in figure 10). This hydrogen bond has also been broken in IM2. Instead, a new hydrogen bond (1.870 Å) to the side chain is created (bond k in figure 12). As a result, the three water molecules now form a hydrogen-bond bridge between the iminol OH and side-chain C=O groups. The energy barrier from IM1 to IM2 is only 0.6 kcal mol−1, and IM2 is lower in energy than IM1 by 0.6 kcal mol−1. Also in IM2, the hydrogen bond involving the iminol OH group (bond b in figure 12) is very short (1.658 Å).
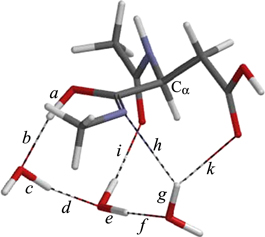
Figure 11. The geometry of TSIM12.
Download figure:
Standard image High-resolution image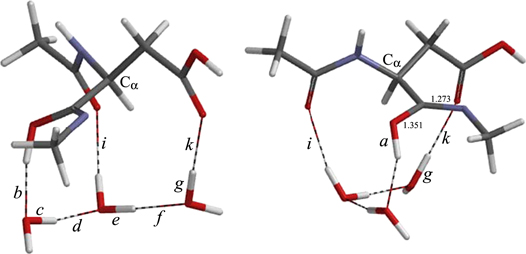
Figure 12. Two views of the geometry of IM2. Bond lengths shown are in Å.
Download figure:
Standard image High-resolution imageNo cyclization TS could be found also from IM2, although the N⋯C distance has been shorten to 2.875 Å. In IM2, the OH group is still 'cis' to the C=N bond; the NCOH dihedral angle is −49° and the distance between the iminol hydrogen and one of the hydrogens of the methyl group is 2.368 Å. We found that the corresponding 'trans' form (IM3, figure 14), where the NCOH dihedral angle is −173°, is lower in energy by about 5 kcal mol−1 than IM2. The conformational change from IM2 to IM3 (i.e., rotation of the iminol OH group) occurs through TSIM23 (figure 13), and the corresponding energy barrier is only 0.4 kcal mol−1. Again, the hydrogen bond involving the iminol OH group in IM3 (bond b in figure 14) is very short (1.655 Å); moreover, the neighboring hydrogen bond in the three-water chain (bond d) is also short (1.657 Å).
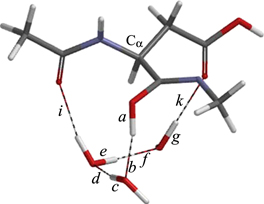
Figure 13. The geometry of TSIM23. The NCOH dihedral angle of the iminol moiety in this geometry is −75°.
Download figure:
Standard image High-resolution image
Figure 14. Two views of the geometry of IM3. Interatomic distances shown are in Å.
Download figure:
Standard image High-resolution imageOn going from IM1 to IM3, ϕ changes from −86° to −148°, and ψ changes from 180° to −115° (while the change in χ1 is modest) (table 1). Such a large change in the main-chain conformation can be a bottleneck of the reaction when the Asp residue resides in a structurally constrained region in a peptide chain.
Cyclization
The N⋯C distance in IM3 is 3.012 Å, which is longer than in IM2. Nevertheless, a cyclization TS (TSIT, figure 15) leading to the gem-diol tetrahedral species (TH, figure 16) has been successfully found from IM3. The energy of TSIT relative to the reactant complex AM is 23.4 kcal mol−1 (see figure 7). This value corresponds to the overall activation barrier for the entire reaction shown in figure 7, and close to the experimental activation energies (25.6 and 21.7 kcal mol−1) for the succinimide formation from TVLDSGISEVR (Aki et al 2013) and VYPDGA (Geiger and Clarke 1987), respectively. The succinimide is formed by dehydration of TH; this process has not been investigated in the present study, because an additional water molecule(s) hydrogen-bonded to the gem-diol group should catalyze it (Catak et al 2009). It may be either that the cyclization is actually the rate-determining step in the succinimide formation, or that the energy of the dehydration TS is comparable to that of the cyclization TS.
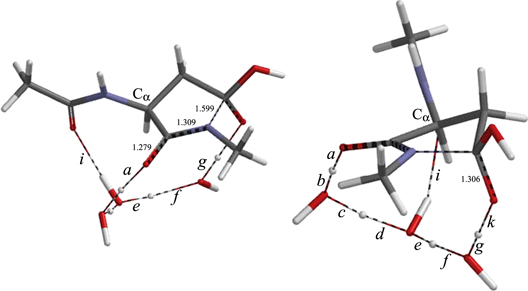
Figure 15. Two views of the geometry of TSIT. Interatomic distances shown are in Å.
Download figure:
Standard image High-resolution image
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 16. Two views of the geometry of TH. Bond lengths shown are in Å.
Download figure:
Standard image High-resolution image{kind=link}
The cyclization results from the nucleophilic attack of the iminol nitrogen on the side-chain carboxyl carbon. Concomitantly with the bond formation between these atoms, a quadruple proton transfer from the iminol OH group to the side-chain C=O group along the three-water bridge and the π-bond reorganization (exchange of single and double bonds) in the OCN moiety occur, resulting in formation of the gem-diol species (TH). In TH, the three water molecules form a hydrogen-bond bridge between the α-C=O group and one of the OH groups of the gem-diol moiety. The N⋯C distance in TSIT is 1.599 Å, and the length of the newly formed N−C bond in TH is 1.482 Å. Changes in the ϕ, ψ and χ1 dihedral angles by 27°, 29° and 36°, respectively, are required in the cyclization step (table 1). The relative energy of TH with respect to the reactant complex AM is 4.2 kcal mol−1.
Conclusions
A novel three-water-assisted mechanism has been proposed for the cyclization reaction of Asp residues leading to the tetrahedral intermediate in the succinimide formation. Key steps are (1) iminolization of the amide group on the C-terminal side and (2) cyclization from the iminol intermediate. Both steps are catalyzed by three water molecules, which act as general acid−base catalysts in proton transfers. The calculated energetics are consistent with experimental results. Therefore, the three-water-assisted mechanism is thought to be one of the possible mechanisms for the Asp cyclization under physiological conditions. A similar mechanism may also be applicable to the succinimide formation from asparagine (Asn) residues in their deamidation reactions (Geiger and Clarke 1987, Stephenson and Clarke 1989, Reissner and Aswad 2003, Robinson and Robinson 2004), and from methylated d-Asp residues enzymatically formed from d-Asp residues (Stadtman 1988, Lowenson and Clarke 1992, Noji et al 2011).