



Abstract
Plugs, i.e. droplets formed in a microchannel, may revolutionize microfluidic cell-based assays. This study describes a microdevice that handles nanolitre-scale liquid plugs for the preparation of various culture setups and subsequent cellular assays. An important feature of this mode of liquid operation is that the recirculation flow generated inside the plug promotes the rapid mixing of different solutions after plugs are merged, and it keeps cell suspensions homogeneous. Thus, serial dilutions of reagents and cell suspensions with different cell densities and cell types were rapidly performed using nanolitres of solution. Cells seeded through the plug processing grew well in the microdevice, and subsequent plug processing was used to detect the glucose consumption of cells and cellular responses to anticancer agents. The plug-based microdevice may provide a useful platform for cell-based assay systems in various fields, including fundamental cell biology and drug screening applications.
Export citation and abstract BibTeX RIS
1. Introduction
Miniaturized systems are promising tools for multiple cell-based assays that reduce reagent consumption and the number of cells required [1–3]. The recent development of lab-on-a-chip and micro total analysis technologies has already provided sophisticated cellular analysis devices [4, 5]. However, in typical microfluidic systems, such as those based on pressure-driven flow, continuous liquid flow has been used, which often leads to non-negligible dead volumes [6, 7]. For example, when a device is connected to external pumps such as microsyringe pumps, despite the small volume required for analysis, i.e. typically less than 1 μl, millilitres of solutions in the lines and syringes will be wasted, which nullifies the advantages of device miniaturization [8]. This mode of operation also complicates handling of multiple solutions because each solution requires a respective microchannel.
A liquid plug is a droplet in a microchannel [9, 10]. The handling of solutions as liquid plugs has a number of advantages over typical continuous liquids: (i) processes require only the necessary amount of solutions (virtually no dead volume), (ii) a series of plugs of different solutions can be processed in a single microchannel and (iii) plugs can be mixed quickly by a plug-specific recirculation flow [11, 12]. These properties have been demonstrated in various fields, including biochemical analysis [13–17]. Especially for cell-based assays, it is important that all necessary operations be performed in a single microdevice. Such operations include preparation of cell suspensions with different cell types and densities, serial dilution of test reagents, sampling of culture medium and mixing of reagents for analysis. A microdevice that handles plugs thus possesses a great potential to conduct such operations on a single chip.
In this study, we describe a microdevice that facilitates the handling of nanolitre liquid plugs for the preparation of cell culture conditions and for the evaluation of metabolic function and responses to anticancer agents. We demonstrate that although the reagent consumption in the microdevice is less than a thousandth of that in conventional instruments, the microdevice can yield comparable results.
2. Materials and methods
2.1. Materials
The cells and reagents used for cell culture experiments were purchased from the following commercial sources: Swiss 3T3 murine fibroblasts (RCB1642) and hepatoblastoma Hep G2 cells (RCB1618) from Riken Cell Bank, Japan; human umbilical vein endothelial cells (HUVEC, CC-2517A), endothelial basal medium-2 (EBM-2, CC-3156), and SingleQuots growth supplement (CC-3162) from Cambrex Bio Science, USA; Dulbecco's modified Eagle medium (DMEM) and foetal bovine serum (FBS) from Invitrogen, USA; fluorescein diacetate and ethidium bromide from Wako, Japan; and DiI and DiO cell-labelling solutions from Invitrogen, USA.
The materials used to fabricate the microdevice were purchased from the following commercial sources: negative photoresist SU-8 2050 from MicroChem Corporation, USA; poly(dimethyl siloxane) (PDMS) from Shin-Etsu Silicones, Japan; and water-repelling fluororesin (Fluorosurf) from Fluorotechnology, Japan. All other chemicals were purchased from Sigma, unless otherwise indicated.
2.2. Device fabrication
The microdevice shown in figure 1 consisted of a glass substrate and a PDMS substrate. The PDMS structures were fabricated using a replica moulding process. Briefly, corresponding master moulds were fabricated with a negative photoresist (SU-8 2050) using photolithography. A PDMS replica of the master was moulded by casting a liquid prepolymer PDMS solution composed of a 10:1 mixture of silicone elastomers and a curing agent. The mixture was cured overnight at room temperature, and the PDMS replica was then peeled off from the master. The PDMS and glass substrates were treated with oxygen plasma and stacked to form microchannels, which created an irreversible bond. The device had a main channel and a handling channel that were connected with a T-junction, as well as 8 cell-culture and measuring regions (figure 1(a)). The ends of all the microchannels were connected to silicone tubes with an inner diameter of 0.5 mm (figure 1(b)). The height of all of the microchannels was 200 μm. The width of the main channel was 600 μm. The width of the culture regions was narrowed to 300 μm to allow the microchannel to precisely place and hold a cell suspension plug (figure 1(c)). A chain of constriction-separated reservoirs was formed in the handling channel, and single reservoirs were introduced in the measuring regions (figure 1(a)). For each reservoir, the narrowest and widest dimensions and volume were set to 150 μm, 450 μm, and 120 nl, respectively. An important requirement for plug operation in a microdevice is that the walls of microchannels must be hydrophobic to stabilize the plugs. Thus, the inner walls of the microchannels were coated with a water-repellent fluororesin, except for the cell culture regions. Prior to cell seeding, the device was filled with 70% (v/v) ethanol for sterilization.
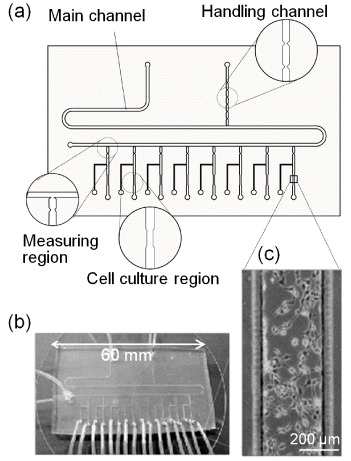
Figure 1. Microdevice for cell-based assays. (a) Schematic with magnified views of the main components, (b) photograph, (c) phase-contrast micrograph of fibroblast cultured in the culture region.
Download figure:
Standard image2.3. Handling of liquid plugs for volume measurements
Liquid plugs were measured, separated, and mixed by controlling the pneumatic pressure at the ends of the microchannels (figure 2). To conduct these operations precisely, constrictions were formed in the handling channel and in the measuring regions, in which the meniscus of a solution spontaneously stopped at the narrowest portion because of the interfacial free energy [14].
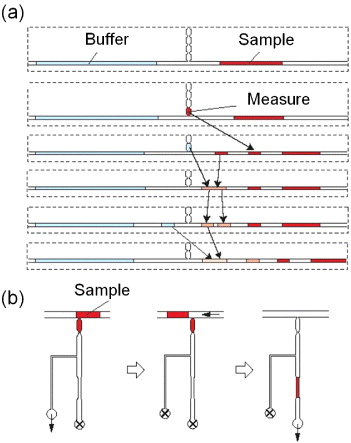
Figure 2. Handling of liquid plugs with the constriction-separated reservoirs (volume: 120 nl). (a) Operations for sequential dilution of a sample with the handling channel. (b) Measurement of a nanolitre plug of cell suspension in the measuring region and seeding of cells in the cell culture region. Arrows indicate the direction of applied pressure and the crossed circles mark closed ends (see movies in [14]).
Download figure:
Standard imageWith this simple structure, serial dilutions of samples were performed as shown in figure 2(a). Briefly, relatively long plugs of sample and buffer solutions (1–2 μl) were introduced into the main channel. A portion of the sample solution was withdrawn into the first reservoir (120 nl) in the handling channel. In this step, plugs of multiple volumes could be prepared by withdrawing the sample solutions into other reservoirs. Subsequently, the solution in the reservoir was separated from that remaining in the main channel and returned to the main channel. In the same manner, a plug of buffer solution was prepared in the main channel. The two plugs were merged, mixed and again divided into two plugs of the same volume using the handling channel. By repeating this process, we prepared a series of solutions with different concentrations of cell suspension and anticancer agents.
2.4. Cell preparation
Fibroblasts and Hep G2 cells were maintained in DMEM supplemented with 10% FBS, 50 units ml−1 penicillin, and 50 μg ml−1 streptomycin. The cells were passaged every 6 days with 0.25% trypsin and 0.02% ethylenediamine tetra-acetic acid. HUVECs were maintained in EBM-2 containing SingleQuots growth supplement. HUVECs from passages 3–8 were used for experiments. The cell passage was conducted every 3–4 days, and the media were changed every other day. All cell cultures were conducted at 37 °C under an atmosphere of 5% CO2 in a humidified incubator.
2.5. Measurement of cell number in the microdevice
Cells were suspended in a culture medium at a density of 1.0 × 106 cells ml−1 for all experiments, unless otherwise indicated, and introduced into the main channel of the microdevice. Serial dilutions of the cell suspension were prepared in the microdevice as described above. When the cell suspension (2.0 × 106 cells ml−1) was directly placed in the cell culture region for viability assays, its volume was initially evaluated using the reservoirs in the measuring regions (figure 2(b)). The density of the seeded cells was quantified by counting the cells in phase-contrast microscopy images of the cell culture region. In addition, the growth of cells in the microdevice was quantified by counting the cells every 12 h after cell seeding for 72 h.
2.6. On-chip measurement of glucose concentration
The glucose consumption by the cells was evaluated and compared with that in a conventional culture dish. Fibroblasts were seeded in the 8-culture chambers at four different cell densities: 1.25 × 105, 2.5 × 105, 5.0 × 105, and 1.0 × 106 cells ml−1. After 6 h of culture for cell adhesion, the culture medium was exchanged. After an additional 12 h of culture, the culture medium was combined with phosphate buffer solution that was previously prepared with the handling channel to dilute the culture medium five-fold. A portion of the diluted sample was withdrawn, separated using the single reservoir in the measuring region, and again diluted by combining it with a phosphate buffer solution to obtain the final 25-fold dilution. A portion of the diluted sample was again measured using the single reservoir and combined with an equal volume of a detection mixture solution containing GOD (400 units ml−1), HRP (100 units ml−1), and AmplexRed (1 mM). GOD catalyzes glucose to hydrogen peroxide and HRP catalyzes hydrogen peroxide and non-fluorescent AmplexRed to fluorescent resorufin. After mixing, fluorescence images were recorded with an IX-71 microscope (Olympus Co., Japan), and the fluorescence intensity was quantified using an image analysis software. The cells in the microdevice were counted, and the number of cells was used to calculate the glucose consumption per cell.
2.7. Drug susceptibility testing with anticancer agents
Hep G2 cells were seeded in the 8-cell culture regions at a density of 1.0 × 106 cells ml−1. After 12 h of culture, the cells were exposed to two anticancer agents: mitomycin C and 5-fluorouracil (5-FU). Three different concentrations of each agent were previously prepared using the handling channel as described above. The plugs were moved to the measuring regions, measured using a single reservoir, and replaced with old culture media in the culture regions. After exposure for 24 h, cell viability was evaluated with a live/dead fluorometric assay using fluorescein diacetate and ethidium bromide [18]. Similar experiments were conducted with conventional culture dishes by preparing the same serial dilutions manually with a pipette.
3. Results and discussion
3.1. Homogeneous cell seeding
Plugs enable efficient handling of solutions and promote large-scale integration. In contrast to continuous fluids, only the necessary amount of solution is necessary and there is virtually no dead volume, which is particularly advantageous for cell-based assays that use valuable biological materials such as human stem cells, growth factors, and lead compounds for drug discovery. In addition, this mode of operation can handle many different solutions in a limited space. Several plugs can be simultaneously moved by applying air pressure from one end of the microchannel. Furthermore, when a plug is moved in a microchannel, recirculation flow occurs inside the plug because of friction between the plug and the microchannel walls, which facilitates the mixing of different solutions after the merging of plugs. This ease of mixing is important because mixing different solutions has been a considerable challenge in conventional microdevices that use continuous flow [19].
The recirculation flow also facilitates the preparation of cell suspension plugs with a uniform cell density because it provides homogeneous suspension. To demonstrate this feature in the fabricated device, we quantified the deviation of cell density for cells seeded at nominally the same density in the 8-cell culture regions in the microdevice. It is well known that cells seeded in a culture dish tend to accumulate in the centre of the dish because centripetal force acts on the cells when the dish is moved and a swirl flow is generated in the culture medium [20, 21]. In this study, there were apparent differences in cell density between the central and peripheral regions in a 35-cm culture dish (figure 3(a)). However, in the microdevice, the density was uniform over the culture regions, even those located at the opposite ends (figure 3(b)). The standard deviation of the cell density in the microdevice was less than one-third of that in the culture dish (figure 3(c)). Note that the dimensions of each cell culture region were designed to be comparable to ∼80% of a 10 × microscope field so that almost all cells in the chamber could be analysed. Nine randomly chosen areas were analysed in a culture dish. Cell density often affects cellular functions such as gene expression and drug sensitivity. Therefore, homogenous cell seeding is crucial for accurate and robust cell-based assays.
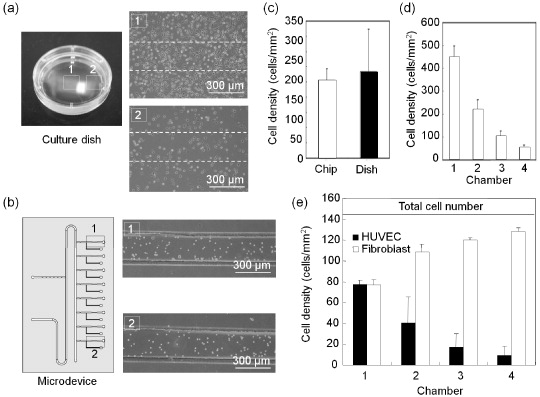
Figure 3. Characterization of cell seeding with plug-based liquid handling. (a) Distribution of the cell density in a culture dish. Representative images around the centre (1) and periphery (2) of a 35-mm culture dish after 12 h of culture. (b) Fibroblasts were introduced into the cell culture regions with the plug-based liquid handling. The inoculated cell density was 200 cells mm−2. (c) Comparison of the cell density distributions in the microdevice and a culture dish. The means and standard deviations were calculated from 24 experiments for the microdevice and nine experiments for the culture dish. (d) Serial dilutions of the cell suspension (density: 2.0 × 106 cells ml−1) processed in the microdevice. The data represent the means and standard deviations from three independent experiments. (e) Preparation of cell suspensions with different compositions of two cell types. The cell densities represent the means and standard deviations from three independent experiments.
Download figure:
Standard image3.2. Preparation of cell suspensions with various cell densities and compositions
A suspension of fibroblasts with a density of 2.0 × 106 cells ml−1 and a culture medium without cells were introduced into the main channel. These two solutions were processed for the preparation of double serial dilutions by repeatedly dividing and merging the two solution plugs with the handling channel (see section 2.3). After the prepared cell suspension plugs were placed in the cell culture regions, the number of cells was counted in microscopy images. The expected decrease in the cell density is shown in figure 3(d). By using a suspension of another cell type instead of a blank culture medium, the same procedure can yield cell suspensions with different compositions of the two cell types. In this study, we initially labelled HUVECs (1.0 × 106 cells ml−1) with DiO (green) and fibroblasts (1.0 × 106 cells ml−1) with DiI (red) to distinguish the cells from each other, and then performed serial dilutions. As shown in figure 3(e), four different compositions of two cell types were prepared, while the total cell number was maintained at almost the same level. Such a setup could be beneficial in biological studies for determining interactions among different types of cells and identifying the roles of individual cell types in complex microenvironments.
3.3. Cell viability and growth
Evaporation is a serious problem in miniaturized devices. Especially for cell culture in nanolitre-scale liquid plugs, evaporation and the associated osmolality shift in the culture medium should be considered. An osmolality shift of above 25% affects cell growth, viability, and secretion [22]. The osmolality shifts were estimated over 24 h by measuring the change in the plug length in the cell culture region. Even in a humidified cell culture incubator, evaporation could not be avoided and ∼40% shift of osmolality was observed after 12 h of incubation. To alleviate this problem, water plugs were placed at both ends of the cell culture region, which was very effective for preventing evaporation, and the osmolality shift was within 20% even after 24 h of incubation. In the following experiments, the culture medium was exchanged every 12 h to adequately minimize the effect of evaporation. The procedures for exchanging culture medium were almost the same as those for cell seeding (Supplemental figure 1 available from stacks.iop.org/STAM/13/064201/mmedia).
A live/dead fluorometric assay was performed to examine cell culture conditions in the microdevice. Fibroblasts were seeded at a relatively high cell density of 2.0 × 106 cells ml−1 and cultured for 48 h. Then, the cells were double-stained with fluorescein diacetate and ethidium bromide. When this assay is performed, the cytoplasm of live cells fluoresces green whereas the nuclei of dead cells emit red light. The assay confirmed that almost all cells were alive (figures 4(a) and (b)). We further examined the culture conditions by measuring the growth of cells. Fibroblasts were seeded at a density of 70 cells mm−2 in the microdevice and a conventional culture dish. The growth of cells was quantified and compared by counting the cells in microscopy images (figure 4(c)). The growth of cells in the microdevice was comparable to that in a culture dish until 48 h. However, a delay of growth was observed after 48 h, probably because of factors such as a shortage of nutrients and a change in pH resulting from the increased number of cells, but not evaporation-related effects. These adverse effects may be reduced by adjusting the ratio of the culture surface area to the volume of the culture medium.

Figure 4. Validation of cell culture conditions in the microdevice. (a, b) Live/dead fluorescent staining of fibroblasts at 48 h of culture in the microsystem. The cytoplasm of live cells fluoresces green (a), whereas the nuclei of dead cells emit red light (b). (c) Comparison of the growth of fibroblasts in the microdevice ( ) and in a culture dish (▪). The data present the means and standard deviations from three independent experiments.
) and in a culture dish (▪). The data present the means and standard deviations from three independent experiments.
Download figure:
Standard image3.4. Glucose consumption assay
To demonstrate that the microdevice can be used not only for cell culture but also for the analysis of metabolic functions, fibroblasts were seeded at four different cell densities. After 12 h of culture, glucose concentration was evaluated by microscopic fluorometry through plug processing (Supplemental figure 2(a)). The final product in the detection reactions was resorufin, which was converted from AmplexRed and yielded strong red fluorescence. In the standard curve for the setup used in this study, a linear relationship between the fluorescence intensity and concentration of resorufin was observed in solutions with a resorufin concentration below 0.1 mM (Supplemental figure 2(b)). Because fresh culture medium contains 5 mM glucose, the sample culture medium had to be diluted up to 50 times to measure the glucose concentration reliably. Therefore, all sample plug solutions were diluted 25 times using the handling channel and subsequently mixed with a plug of the detection reaction solution at a volume ratio of 1:1 to dilute the sample 50 times. The glucose consumption rate per cell was decreased by increasing the cell density (table 1). Considering that 72% of the glucose was already consumed in the relatively short culture period at the highest cell density used in this study, the observed concentration-dependent consumption may be reasonable. In a conventional culture dish, the glucose consumption rates were higher than those in the microdevice, especially at the highest seeding cell density (table 1). This is probably due to the differences in oxygen supply. Considering that oxygen solubility in PDMS is very high, 10 times greater than that in the culture medium [23, 24], and the height of the culture regions was 200 μm whereas the thickness of culture medium in a conventional culture dish was ∼2 mm, more efficient ATP production through aerobic metabolism is expected in the microdevice.
Table 1. Glucose consumption of fibroblasts during 12 h of culture.
| Seeding cell density (×105 cell ml−1) | Glucose concentration* (mM) | Glucose consumption rate (pmol 12 h−1 cell−1) | ||
|---|---|---|---|---|
| Microdevice | Dish | Microdevice | Dish | |
| 1.25 | 3.77 ± 0.20 | 4.34 ± 0.08 | 9.84 ± 1.60 | 10.05 ± 1.26 |
| 2.5 | 3.38 ± 0.24 | 3.94 ± 0.18 | 6.48 ± 0.96 | 8.45 ± 1.42 |
| 5.0 | 2.56 ± 0.32 | 3.46 ± 0.20 | 4.88 ± 0.64 | 6.17 ± 0.81 |
| 10 | 1.40 ± 0.15 | 1.28 ± 0.25 | 3.60 ± 0.15 | 7.40 ± 1.42 |
3.5. Cytotoxicity assays with anticancer agents
Drug susceptibility testing in a miniaturized system could be useful for the simultaneous screening of a large number of small-volume drugs, especially when using precious cells such as the primary cells obtained from a cancer patient through biopsy. A series of plugs containing two anticancer agents with three different concentrations was prepared in the microdevice, and Hep G2 cells were exposed to the agents for 24 h. For comparison, the same experiments were conducted with conventional culture dishes. A similar tendency was observed using both agents. The viability of cells decreased linearly when the concentration of the agents increased (figures 5(a) and (b)). These results are similar to those observed with conventional culture dishes. In addition, the results for mitomycin C were consistent with those given in previous reports in which the IC50 was 6–9 μg ml−1 [25, 26]. Although the consumption of the agents and cells in the microdevice is less than a thousandth of that in conventional instruments, the microdevice can be used to obtain comparable results.
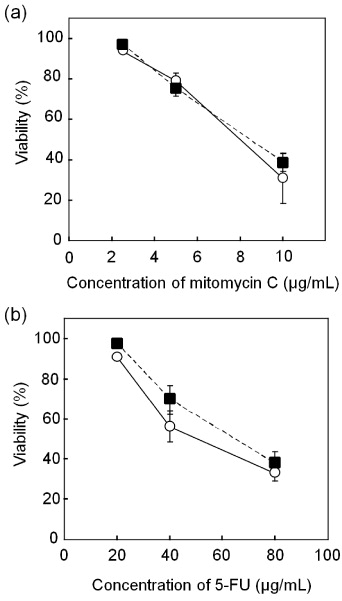
Figure 5. Susceptibility testing for two anticancer drug agents. After 12 h of culture, Hep G2 cells were exposed to the anticancer agents for 24 h, and their viability was quantified in the microdevice ( ) and in a culture dish (▪). Responses to mitomycin C (a) and 5-FU (b). The data present the means and standard deviations from two independent experiments.
) and in a culture dish (▪). Responses to mitomycin C (a) and 5-FU (b). The data present the means and standard deviations from two independent experiments.
Download figure:
Standard image4. Conclusions
In the development of a lab on a chip, the handling of solutions as a continuous liquid flow has hindered operations generally conducted in a cell biology laboratory such as measurement, separation and mixing of solutions. Here we proposed a plug-based liquid-handling approach that enabled multiple operations on a single chip without dead volume. The plug-specific recirculation flow permitted serial dilution of the cell suspension and homogeneous cell seeding in the microdevice fabricated in this study. Cells cultured in the microdevice showed a favourable survival and growth behaviour. Comparable data were obtained for glucose consumption and drug susceptibility against anticancer agents with the microdevice, while using the volumes of reagents and cells less than a thousandth of those required in a conventional dish.
Acknowledgments
This study was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas, MEXT, Japan, a Health Labour Sciences Research Grant, MHLW, Japan, and the Industrial Technology Research Grant Program from NEDO, Japan.