



Abstract
The conductances of molecular model junctions comprising a triazatriangulenium platform with or without an ethynyl spacer and an upright Zn-porphyrin are probed with a low-temperature scanning probe microscope. The platform alone is found to be highly conductive. The ethynyl-linked Zn-porphyrin moiety reduces the conductance by three orders of magnitude and leads to an unexpected, non-monotonous variation of the force that was measured simultaneously at the tip of the microscope. Density functional theory calculations show that this variation results from an induced tilting of the porphyrin.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Scanning tunneling microscopy (STM) is useful for probing the conductance of single molecules because it enables a detailed characterization of the junction via imaging of the molecule prior to and after the conductance measurement, in particular at low-temperature and under ultra-high vacuum conditions. Given the sensitivity of the conductance with respect to atomic distances, the forces between the electrodes and a molecule and their variation with the electrode separation are of great interest. Nevertheless, the number of studies combining conductance and force measurements on molecules using STM combined with non-contact atomic force microscopy (AFM) is limited. Planar molecules [1–3], fullerenes [4–6], and carbonmonoxide [7] have been investigated. To implement advanced functions at the nanoscale, it is desirable to rationally design molecules that are more complex [8, 9]. Here we address a generic model (Por-TATA, figure 1(a)) comprising the molecular platform tri-octyl-triazatriangulenium (tri-octyl-TATA) [10], an intervening spacer (ethynyl), and a functional unit (Zn-porphyrin). The molecular complex was designed to attach functional molecules in an upright and free-standing position onto a surface as well as to ensure a high conductivity due to conjugated bonds within the platform, the spacer, and the Zn-porphyrin. STM and simultaneous AFM are used to characterize the conductance and the forces at the junction. To make these complex molecules accessible to measurements in a controlled ultra-high vacuum, low-temperature environment a home-built electrospray ionization source is used. The experiments are complemented by calculations of the electron transport and the forces.

Figure 1. (a) Lewis structure of Zn-porphyrin-functionalized tri-octyl-triazatriangulenium (Por-TATA). (b) Mass spectrum of sprayed molecules. (c) STM image of tri-octyl-TATA, Zn-porphyrin, and Por-TATA (80 mV, 9 pA,  2). (d) Single Por-TATA with an overlaid molecular sketch with a perpendicular porphyrin attached to the platform (
2). (d) Single Por-TATA with an overlaid molecular sketch with a perpendicular porphyrin attached to the platform ( , 13 pA,
, 13 pA,  2). (e) Scanning tunneling spectrum of the affinity and ionization levels recorded at the center position of the imaged Por-TATA (protrusion in (d)). A gap of 2.80(5) eV
2). (e) Scanning tunneling spectrum of the affinity and ionization levels recorded at the center position of the imaged Por-TATA (protrusion in (d)). A gap of 2.80(5) eV  270(5) kJ mol−1 is estimated from the positions of the maxima (dashed lines).
270(5) kJ mol−1 is estimated from the positions of the maxima (dashed lines).
Download figure:
Standard image High-resolution imageThe experimental data reveal that the platform itself exhibits a high conductance making it a suitable linker for conductive molecules. On the Por-TATA molecule, we observed unusual non-monotonous variation of the force which also leaves a footprint on the conductance. A comparison with the results of quantum chemical calculations shows that a bending of the molecule occurs as the tip approaches the porphyrin moiety.
2. Experiment
Experiments were performed in a combined STM/AFM in UHV at 5 K with a base pressure of  . The AFM was operated in a non-contact frequency-modulation mode with a tuning fork probe [11]. The free prong oscillated at a constant amplitude of
. The AFM was operated in a non-contact frequency-modulation mode with a tuning fork probe [11]. The free prong oscillated at a constant amplitude of  Å at its resonance frequency of
Å at its resonance frequency of  . A W-tip was attached to the free prong to simultaneously measure the conductance along with the frequency shift (see supplementary material, section 2 for more details). The differential conductance was measured with a lock-in amplifier and with the current feedback loop disabled. All STM images were acquired in constant-current mode with the bias voltage V applied to the sample. The W-tip was prepared in situ by repeatedly indenting into the Au substrate until it was covered with surface material. Au(111) surfaces were cleaned by Ar+ sputtering and annealing cycles. The synthesis of Por-TATA has been presented in [12]. Por-TATA was sprayed from a
. A W-tip was attached to the free prong to simultaneously measure the conductance along with the frequency shift (see supplementary material, section 2 for more details). The differential conductance was measured with a lock-in amplifier and with the current feedback loop disabled. All STM images were acquired in constant-current mode with the bias voltage V applied to the sample. The W-tip was prepared in situ by repeatedly indenting into the Au substrate until it was covered with surface material. Au(111) surfaces were cleaned by Ar+ sputtering and annealing cycles. The synthesis of Por-TATA has been presented in [12]. Por-TATA was sprayed from a  mM chloroform/methanol (1:1) solution in the positive ion mode using an in situ ultra-high vacuum electrospray ionization setup [13]. Before deposition, the molecular beam was analyzed by mass spectrometry. During deposition the Au(111) sample was kept in the preparation chamber at a pressure of 2 × 10−9 mbar over a period of
mM chloroform/methanol (1:1) solution in the positive ion mode using an in situ ultra-high vacuum electrospray ionization setup [13]. Before deposition, the molecular beam was analyzed by mass spectrometry. During deposition the Au(111) sample was kept in the preparation chamber at a pressure of 2 × 10−9 mbar over a period of  h. An ion impact energy of 8(4) eV was used.
h. An ion impact energy of 8(4) eV was used.
3. Theoretical methods
Simulations of force and conductance curves of a platform-mounted Zn-porphyrin molecule were performed by combining constrained molecular structure optimizations using static Kohn–Sham density functional theory (KS-DFT) [14] and the Landauer–Imry–Büttiker approach [15, 16]. The optimization of molecular structures was carried out using the quantum chemical program package TURBOMOLE 6.0 [17] with the pure exchange-correlation functional BP86 [18, 19] and Ahlrichs' split-valence triple-zeta basis set with polarization functions on all atoms (def-TZVP) [20, 21]. Dispersion interactions were taken into account via Grimmeʼs empirical correction scheme DFT-D2 [22]. In Turbomole 6.0 no C6 parameters for gold are available. The convergence criteria were 5 × 10−4 kJ mol−1 Å−1 (10−7 a.u.) for the energy in the self-consistent field algorithm and 0.5 kJ mol−1 Å−1 (10−4 a.u.) for the gradient in molecular structure optimizations. The transmission functions, the conductance at zero-voltage, and central subsystem MOs were calculated using the post-processing tool artaios [23, 24]. In order to obtain the overlap and Fock matrices, single-point calculations using the gaussian 09 quantum chemistry program package [25] with the hybrid functional B3LYP and the Los Alamos (LANL2DZ) effective core potentials [26] were employed. A small basis set was chosen to reduce ghost transmission [23]. The B3LYP functional [27, 28] was chosen to bypass convergence problems with BP86. Further details on the calculation are provided in appendix
4. Characterization of Por-TATA
Molecular Por-TATA (figure 1(a)) ions sprayed from a chloroform/methanol solution were analyzed by mass spectrometry prior to deposition on the surface. The spectrum in figure 1(b) reveals singly charged, intact Por-TATA (1014 u/e) as well as tri-octyl-TATA [8, 9, 29] molecules (618 u/e). The partial fragmentation is most likely due to a protonation of the molecule during spraying, which destabilizes the bond between tri-octyl-TATA and Zn-porphyrin. The resulting tri-octyl-TATA cation as well as the 5-ethynyl-Zn-porphyrin are highly stable species. No peak corresponding to 5-ethynyl-Zn-porphyrin is observed, which indicates a low ionization cross-section.
The STM image in figure 1(c) is typical of the structures observed after deposition of Por-TATA on the Au surface. From an analysis of many surface areas we find that only  % of the adsorbed molecules are intact Por-TATA. We attribute the fragmentation during adsorption to the impact and adsorption energies [30]. In addition, excitation of molecular ions may occur during their passage through the mass spectrometer [31]. The STM image (figure 1(c)) shows a cluster of one Por-TATA and two tri-octyl-TATA molecules as well as a single Zn-porphyrin. The latter is readily identified owing to its four-fold symmetry [32]. The TATA platforms exhibit an expected threefold symmetry as reported in [10]. At the boundary of the cluster octyl groups (oct) oriented either clockwise or anti-clockwise are discernible [10]. Intact Por-TATA (figures 1(c) and (d)) is signaled by a protrusion at the center of the platform. This interpretation was tested by applying elevated voltages to a related molecule (Por-TATA with three phenyl groups attached to the meso positions of the porphyrin) to induce fragmentation (figure S1 in the supplementary material). After this procedure, tri-octyl-TATA and nearby Zn-porphyrin were observed. In addition, scanning tunneling spectra were recorded above the intact Por-TATA (figure 1(e)). While the relative intensity of the two molecular resonances may vary on different molecules, the data reveal an energy gap4
of
% of the adsorbed molecules are intact Por-TATA. We attribute the fragmentation during adsorption to the impact and adsorption energies [30]. In addition, excitation of molecular ions may occur during their passage through the mass spectrometer [31]. The STM image (figure 1(c)) shows a cluster of one Por-TATA and two tri-octyl-TATA molecules as well as a single Zn-porphyrin. The latter is readily identified owing to its four-fold symmetry [32]. The TATA platforms exhibit an expected threefold symmetry as reported in [10]. At the boundary of the cluster octyl groups (oct) oriented either clockwise or anti-clockwise are discernible [10]. Intact Por-TATA (figures 1(c) and (d)) is signaled by a protrusion at the center of the platform. This interpretation was tested by applying elevated voltages to a related molecule (Por-TATA with three phenyl groups attached to the meso positions of the porphyrin) to induce fragmentation (figure S1 in the supplementary material). After this procedure, tri-octyl-TATA and nearby Zn-porphyrin were observed. In addition, scanning tunneling spectra were recorded above the intact Por-TATA (figure 1(e)). While the relative intensity of the two molecular resonances may vary on different molecules, the data reveal an energy gap4
of  as expected for Zn-porphyrin [33, 34]. This agreement between the energy gap of Por-TATA and Zn-porphyrins suggests that the molecular orbitals of the porphyrin are only weakly perturbed by orbitals of the platform and metal surface. The measured energy of the molecular resonance associated with the lowest unoccupied molecular orbitals (LUMO, at 1.6 V) is significantly different from those measured on the fragments Zn-porphyrin and tri-octyl-TATA (figure S3 of the supplementary material).
as expected for Zn-porphyrin [33, 34]. This agreement between the energy gap of Por-TATA and Zn-porphyrins suggests that the molecular orbitals of the porphyrin are only weakly perturbed by orbitals of the platform and metal surface. The measured energy of the molecular resonance associated with the lowest unoccupied molecular orbitals (LUMO, at 1.6 V) is significantly different from those measured on the fragments Zn-porphyrin and tri-octyl-TATA (figure S3 of the supplementary material).
The gap calculated by DFT (B3LYP, 2.67 eV) between the highest occupied and the lowest unoccupied molecular orbitals (HOMO and LUMO, figure 2(a)) of the Por-TATA on a Au monolayer agrees reasonably well with the experimental result. This value is only slightly larger than the calculated gap of isolated Por-TATA using the same exchange-correlation functional with dispersion correction (2.60 eV, section 7 in the supplementary material). Although quantitative differences occur using a pure functional (BP86-D), the MO energies are qualitatively in the same order. Furthermore, the HOMO–LUMO gaps of isolated Por-TATA fragments, e.g. ethynyl-TATA and ethynyl-Zn-porphyrin, are significantly larger than for the intact Por-TATA, which supports the interpretation of intact Por-TATA molecules (figure S7).

Figure 2. (a) Isosurface plots (isovalue 0.02) of the frontier central subsystem molecular orbitals of Por-TATA. (b) Potential energy hypersurface scan between straight and bent Por-TATA structures with a pyramid-shaped Au tip (relative distance between tip and molecule  Å). d was chosen such that the difference in energy for the two structures is minimal. The abscissa shows a linear interpolation between straight and bent Por-TATA structures generated by a linear synchronous transit method using internal coordinates.
Å). d was chosen such that the difference in energy for the two structures is minimal. The abscissa shows a linear interpolation between straight and bent Por-TATA structures generated by a linear synchronous transit method using internal coordinates.
Download figure:
Standard image High-resolution imageThe roundish shape of Por-TATA (figure 2(d)) suggests that the porphyrin may rotate around the ethynyl linker. This is consistent with the calculated rotation barrier of Zn-porphyrin on tri-octyl-TATA of 1.3 kJ mol−1 (figure S4). If Zn-porphyrin was rotationally restricted, an elongated shape would be expected similar to upright coronene [35].
Closer inspection of images of single Por-TATA molecules (figure 1(d)) shows that the Zn-porphyrin is not centered on the tri-octyl-TATA platform. This is likely due to a deformation of the molecule in the presence of the STM tip. From an interpolation between a straight and a bent structure (obtained from constrained molecular structure optimizations, appendix
5. Contact formation to Por-TATA
To investigate the contact formation between a single Por-TATA molecule and the STM tip the short-range force5
Fs and conductance G are measured simultaneously with respect to the tip-molecule distance (figure 3(a), circles and squares). The tip was initially retracted by 1.5 nm from the molecule to prevent a tilt due to the influence of the tip. As the tip is brought closer again, a shallow minimum of the short-range force Fs (arrow 1 in figure 3(a)) occurs with a maximal attractive force  . According to our DFT calculations (vide infra), this corresponds to the tip contacting one of the topmost hydrogen atoms or, as the molecule may be slightly tilted, the upper carbon atoms of the Zn-porphyrin macrocycle6
. As the tip further approaches the molecule, the force initially becomes less attractive as expected. However, subsequently the attraction increases again to large values. Further reduction of the tip–molecule distance beyond the range shown in figure 3(a) lead to damage of the molecule or the tip.
. According to our DFT calculations (vide infra), this corresponds to the tip contacting one of the topmost hydrogen atoms or, as the molecule may be slightly tilted, the upper carbon atoms of the Zn-porphyrin macrocycle6
. As the tip further approaches the molecule, the force initially becomes less attractive as expected. However, subsequently the attraction increases again to large values. Further reduction of the tip–molecule distance beyond the range shown in figure 3(a) lead to damage of the molecule or the tip.
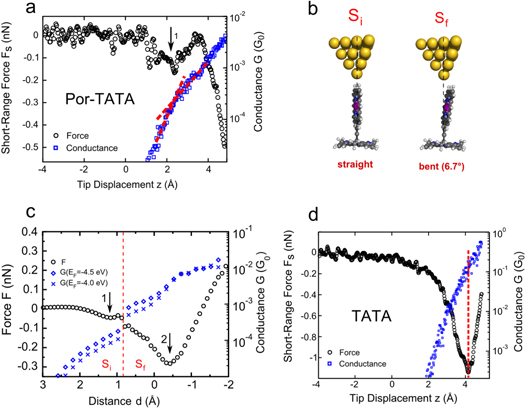
Figure 3. (a) Simultaneously measured short-range force (circles, left axis) and conductance (squares, right axis) on a Por-TATA at the location of the Zn-porphyrin (with 0.1 V applied to the sample). The arrow indicates the shallow force minimum. (b) Snapshots of constrained-optimized model structures (the Por-TATA with a pyramid-shaped Au tip) for the structures Si (straight) and Sf (bent by 6.7°) with the tilt angle defined according to figure A1(c). (c) Calculated force (left axis) and for ghost transmission corrected conductance (right axis) as a function of the relative tip–molecule distance for different values of the Fermi energy. The dashed line indicates the change of the minimum-energy, molecular structure (from Si to Sf). Arrows mark force minima 1 and 2. d = 0 Å was set to the structure obtained from an unconstrained optimization of the tip–molecule system (Au–H internuclear distance of 1.3 Å). (d) Simultaneously measured force (circles, left axis) and conductance (squares, right axis) on a tri-octyl-TATA molecule at V = 50 mV.
Download figure:
Standard image High-resolution imageThe simultaneously measured conductance G(z) increases monotonously. At the point of maximal attraction, we determine a contact conductance  (with
(with  the conductance quantum, e: elementary charge, and h: Planckʼs constant). The contact conductance is within the range (
the conductance quantum, e: elementary charge, and h: Planckʼs constant). The contact conductance is within the range ( G0) measured for a similar junction (Zn-porphyrin with two thiolphenyl linkers) using mechanical break-junction techniques [36–38].
G0) measured for a similar junction (Zn-porphyrin with two thiolphenyl linkers) using mechanical break-junction techniques [36–38].
We first discuss the surprising observation of two attractive wells. It might be linked to plastic deformations of the junction. However, no corresponding increase of the dissipation of the tuning fork was observed. Furthermore, data recorded during approach and retraction of the tip show no hysteresis, which indicates an elastic deformation. An alternative explanation for force variation is a slight bending or tilting of the Zn-porphyrin that occurs when the interaction with the tip apex atom becomes repulsive at  . This idea is further developed using DFT calculations to simulate force and conductance measurements of a platform-mounted Zn-porphyrin molecule. The tip was placed above the middle H-atom of the Zn-porphyrin of a straight Por-TATA molecule (figure 3(b), structure Si). Next, the tip–molecule distance d was decreased in a stepwise fashion and the structures were optimized while holding selected atoms of the tip and the TATA platform fixed (see figure A1(a)). When d is reduced the structure Si undergoes an abrupt change to a more bent shape Sf. This change corresponds to the transition from the left to the right minimum in figure 2(b), which is presumably less abrupt in the experiment due to fluctuations of the molecule. For smaller d the energy of the right minimum is expected to shift to lower energies than the left minimum due to strong repulsive tip–molecule interactions. Figure 3(c) shows the calculated forces and conductances. The dashed line indicates the positions where abrupt bending occurred (d = 0.9 Å). The force exhibits two minima (arrows 1 and 2) that correspond to maximal attraction. The first, shallow maximum occurs when the tip contacts the middle H-atom of the porphyrin ring while the second one reflects the interaction of the Au tip with carbon atoms from the porphyrin macrocycle (figure A1(b)). As the barrier between a straight and a bent Por-TATA is below the estimated error margin of the DFT calculations (0.1 kJ mol−1, figure 2(b)), the calculations were also performed for a bent Por-TATA as starting structure, which virtually leads to the same results (appendix
. This idea is further developed using DFT calculations to simulate force and conductance measurements of a platform-mounted Zn-porphyrin molecule. The tip was placed above the middle H-atom of the Zn-porphyrin of a straight Por-TATA molecule (figure 3(b), structure Si). Next, the tip–molecule distance d was decreased in a stepwise fashion and the structures were optimized while holding selected atoms of the tip and the TATA platform fixed (see figure A1(a)). When d is reduced the structure Si undergoes an abrupt change to a more bent shape Sf. This change corresponds to the transition from the left to the right minimum in figure 2(b), which is presumably less abrupt in the experiment due to fluctuations of the molecule. For smaller d the energy of the right minimum is expected to shift to lower energies than the left minimum due to strong repulsive tip–molecule interactions. Figure 3(c) shows the calculated forces and conductances. The dashed line indicates the positions where abrupt bending occurred (d = 0.9 Å). The force exhibits two minima (arrows 1 and 2) that correspond to maximal attraction. The first, shallow maximum occurs when the tip contacts the middle H-atom of the porphyrin ring while the second one reflects the interaction of the Au tip with carbon atoms from the porphyrin macrocycle (figure A1(b)). As the barrier between a straight and a bent Por-TATA is below the estimated error margin of the DFT calculations (0.1 kJ mol−1, figure 2(b)), the calculations were also performed for a bent Por-TATA as starting structure, which virtually leads to the same results (appendix
The calculated forces reproduce the essential features of the experimental force data. They exhibit a similar shallow attractive well and are also consistent with the growing attraction at shorter distances. The transport calculations showed an artificially high transmission in the energy range of the expected Fermi energy caused by the so called 'ghost transmission' ([23] and supplementary material, section 9). After subtracting the ghost transmission the calculated conductances are similar to the experimental results. We verified that the calculated conductances are independent of the choice of the Fermi energy  over the range from
over the range from  to
to  . Moreover, no transmission peaks are present near the Fermi energy for all examined tip–molecule distances (figure S8), which suggests that the Landauer formalism is a good description of electron transport through the junction [39]. We note that the quantitative agreement of the conductances observed at the position of the first attractive force maximum may be due to a favorable cancellation of errors.
. Moreover, no transmission peaks are present near the Fermi energy for all examined tip–molecule distances (figure S8), which suggests that the Landauer formalism is a good description of electron transport through the junction [39]. We note that the quantitative agreement of the conductances observed at the position of the first attractive force maximum may be due to a favorable cancellation of errors.
To estimate the contribution of the TATA platform to the resistance of the Por-TATA molecule we repeated the experiments on the platform alone. Figure 3(d) shows force (circles) and conductance (squares) data recorded above the center of a TATA molecule. Compared to Por-TATA the maximal attractive force is larger by an order of magnitude. This is expected because the tip gets closer to the platform, which increases the dispersion force. At the force minimum (dashed line in figure 3(d)) the conductance is  G0. DFT calculations (figure S10) predict a conductance of
G0. DFT calculations (figure S10) predict a conductance of  G0 in reasonable agreement with the measured value. This high value comes close to that of C
G0 in reasonable agreement with the measured value. This high value comes close to that of C [40]. In other words, the TATA platform is a highly conductive linker to metal surfaces.
[40]. In other words, the TATA platform is a highly conductive linker to metal surfaces.
In the Landauer model of coherent transport the total transmission is the product of the transmissions of the subunits [41]. This implies that the contact conductance of Por-TATA ( ) may be attributed to the combined effects of the platform (
) may be attributed to the combined effects of the platform ( ) and the Zn-porphyrin together with an ethynyl spacer (
) and the Zn-porphyrin together with an ethynyl spacer ( ) as estimated from a calculation on an ethynyl thiolate spacer (figure S10).
) as estimated from a calculation on an ethynyl thiolate spacer (figure S10).
6. Conclusion
In conclusion, single-molecule junctions of Zn-porphyrin functionalized tri-octyl-triazatriangulenium were prepared in a low-temperature STM/AFM. The molecular platform leads to an almost upright orientation of the Zn-porphyrin moiety. The ethynyl spacer reduces the electronic coupling between the Zn-porphyrin and the surface as shown by tunneling spectroscopy. Measurements and DFT calculations show that the presence of the tip induces a tilting of the porphyrin. Force and conductance curves on the platform alone show that it is a highly conductive molecular linker to metal surfaces.
Acknowledgments
Financial support from the Fonds der Chemischen Industrie and the Deutsche Forschungsgemeinschaft via the SFB 677 is gratefully acknowledged. We thank the University of Hamburg High Performance Computing Centre and the North-German Centre for High-Performance Computing (HLRN) for computational resources.
Appendix A.: Theoretical details
In general, either static DFT calculations or molecular dynamic (MD) simulations [42] are performed to simulate STM and AFM processes. MD simulations provide richer information but are typically much more computationally demanding (for a given method of calculating energies and forces). In order to address the unexpected flexibility of the molecule, MD simulations may achieve a comprehensive understanding. However, since the experiments have been performed at time scales up to one minute, MD simulations will not correctly reproduce the behavior of the system due to a sliding speed that has to be larger by several orders of magnitude due to practical limitations on simulation time. Therefore, we performed constrained optimizations using static DFT.
A.1. Structure optimization
The tip was modeled by a pyramid consisting of 20 gold atoms. The octyl chains attached to the nitrogen atoms were replaced with methyl groups (unless otherwise mentioned) to reduce computation times. This substitution does not modify the electronic structure of TATA (section 8 in the supplementary material).
A series of structures consisting of the Por-TATA and the 20 atom gold tip were optimized first with no constraint and subsequently with the constraint that the coordinates of three hydrogen atoms of the TATA platform and the topmost slab of the tip are held fixed (figure A1(a), marked in red). The structure obtained from an unconstrained optimization has an internuclear Au–H distance of  Å. Note that the distance between the Au and H nuclei
Å. Note that the distance between the Au and H nuclei  is not the same as the tip–molecule distance d (figure A1(b)). For the constrained optimizations and subsequent force calculations the gold surface was neglected (contrary to the electron transport calculations).
is not the same as the tip–molecule distance d (figure A1(b)). For the constrained optimizations and subsequent force calculations the gold surface was neglected (contrary to the electron transport calculations).

Figure A1. (a) Constrained-optimized Por-TATA with a 20 atom gold tip using DFT(BP86-D)/def-TZVP. The fixed hydrogen and gold atoms are circled in red. (b) Constrained-optimized structures Si (straight) at d = 0.00 Å and Bf (strongly bent, (14.7°)) at  Å. Different distances defined as Au–H distance (
Å. Different distances defined as Au–H distance ( ) (red) and tip–molecule distance d (blue) are illustrated using both structures as an example. In the case of structure Bf, the tip also interacts with the non-hydrogen atoms of the porphyrin macrocycle. (c) Tilt angle α between the porphyrin planes of structure type Si and structure type Sf (bent by 6.7°) at a tip–molecule distance d = 0.7 Å. (d) Internuclear distances between the top Au atom of the tip and the contacted H-atom plotted against the tip–molecule distance for different configurations of the types Si and Sf (figure 3(b)) and Bi, Bf (figure A2(a)). The tip–molecule distance d = 0.00 Å corresponds to the equilibrium structure, see straight Por-TATA structure in (b).
) (red) and tip–molecule distance d (blue) are illustrated using both structures as an example. In the case of structure Bf, the tip also interacts with the non-hydrogen atoms of the porphyrin macrocycle. (c) Tilt angle α between the porphyrin planes of structure type Si and structure type Sf (bent by 6.7°) at a tip–molecule distance d = 0.7 Å. (d) Internuclear distances between the top Au atom of the tip and the contacted H-atom plotted against the tip–molecule distance for different configurations of the types Si and Sf (figure 3(b)) and Bi, Bf (figure A2(a)). The tip–molecule distance d = 0.00 Å corresponds to the equilibrium structure, see straight Por-TATA structure in (b).
Download figure:
Standard image High-resolution imageA.2. Transmission and molecular orbitals
For the calculation of the transmission the methyl-TATA platform of the isolated, optimized Por-TATA was replaced by a platform (tri-octyl-TATA) that was previously optimized on four layers of Au (DFT(PBE-D)/SVP) [10]. Additionally, the Por-TATA was hosted on a single Au layer consisting of 20 atoms with N–Au distances of 3.2 Å as optimized in [10]. Details on the electron transport calculations as well as Cartesian coordinates are given in sections 5 and 11 of the supplementary material.
The resulting finite-cluster system was divided into tip (left), surface (right), and Por-TATA molecule (central) subsystem (supplementary material, figure S5). All gold atoms were assigned to the electrodes. Thus, the definition of the central subsystem comprises only the Por-TATA molecule.
The central subsystem MOs of the Por-TATA were calculated by solving the central subsystemʼs secular equations and determined by comparing its shape with the MOs of the isolated Por-TATA. Increasing the basis set to triple-zeta quality (def-TZVP) for the isolated Por-TATA did not result in significant changes in the energy gaps (supplementary material, section 7). The MOs were visualized with MOLDEN [43] and a local postprocessing tool.
A.3. Calculated contact curves
The force was approximated as the negative of the numerical derivative of the energy values with respect to the tip–molecule distance d, which represents the displacement along the tip–TATA platform direction. d is defined as the height of the whole tip–Por-TATA system (distance between the highest coordinate of the tip and the lowest coordinate of the Por-TATA) minus the height of the fully optimized structure that corresponds to d = 0.00 Å, figure A1(b).
The constrained optimizations using DFT(BP86-D)/def-TZVP were started from structure Si (straight, figure 3(b)). The tip–molecule distance d was elongated and reduced by steps of 0.20 Å over a range of 5.00 Å, starting with an elongation of 3.00 Å. For distances  Å (see abscissa in figures 3(c) and A1(d)) the porphyrin position remains qualitatively unchanged after the relaxation. For smaller distances the Por-TATA tilts and the lowest energy structure changes to the bent structure Sf (figure 3(b),
Å (see abscissa in figures 3(c) and A1(d)) the porphyrin position remains qualitatively unchanged after the relaxation. For smaller distances the Por-TATA tilts and the lowest energy structure changes to the bent structure Sf (figure 3(b),  with α defined in figure A1(c)). Upon further approach of the tip, the Por-TATA structure is even more bent (
with α defined in figure A1(c)). Upon further approach of the tip, the Por-TATA structure is even more bent ( for a tip–molecule distance of
for a tip–molecule distance of  Å). It is to be noted that the bent structure does not converge to the straight structure when used as a starting structure (even for large tip–molecule distances), which can be observed in calculations without the Au tip as well (see figure 2(b) and figure S6, respectively).
Å). It is to be noted that the bent structure does not converge to the straight structure when used as a starting structure (even for large tip–molecule distances), which can be observed in calculations without the Au tip as well (see figure 2(b) and figure S6, respectively).
However, using the bent structure ( ) as starting structure for the constrained optimizations leads to energetically more favorable converged structures for distances
) as starting structure for the constrained optimizations leads to energetically more favorable converged structures for distances  Å (figure 3(c)) compared with the straight structure. Therefore, structures of the type Si (straight) are used for distances
Å (figure 3(c)) compared with the straight structure. Therefore, structures of the type Si (straight) are used for distances  Å and structures of the type Sf (
Å and structures of the type Sf ( ) as initial guess for smaller distances for the calculations shown in figure 3(c), in which this structure is consistently more stable than the straight one.
) as initial guess for smaller distances for the calculations shown in figure 3(c), in which this structure is consistently more stable than the straight one.
The same procedure has been applied for the structures Bi ( ) and Bf (
) and Bf ( , figure A2(b)), which virtually results in the same shape of the force and conductance curves than the calculations shown in figure 3(c).
, figure A2(b)), which virtually results in the same shape of the force and conductance curves than the calculations shown in figure 3(c).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure A2. (a) Snapshots of constrained-optimized model structures (the Por-TATA with a pyramid-shaped Au tip) Bi ( °) and Bf (
°) and Bf ( °) with the tilt angle defined according to figure A1(c). (b) Calculated force (left axis) and conductance (right axis) as a function of the relative tip–molecule distance for different values of the Fermi energy. The conductance is corrected for ghost transmission. The dashed line indicates the change of the minimum-energy, molecular structure (from Bi to Bf). Arrows mark force minima 1 and 2.
°) with the tilt angle defined according to figure A1(c). (b) Calculated force (left axis) and conductance (right axis) as a function of the relative tip–molecule distance for different values of the Fermi energy. The conductance is corrected for ghost transmission. The dashed line indicates the change of the minimum-energy, molecular structure (from Bi to Bf). Arrows mark force minima 1 and 2.
Download figure:
Standard image High-resolution image{kind=link}
Figure A1(d) illustrates the sudden increase of the distance between the tip atom and the middle H-atom of the porphyrin ring as the molecule slips past the tip. This sudden increase of d causes the sharp kink in the conductance curve at d = 0.80 Å (figure 3(c)) and d = 0.50 Å (figure A2(b)).
Footnotes
- 4
The conductance gap may appear larger than the HOMO–LUMO gap if the applied voltage does not entirely drop over the vacuum gap. In the present case, however, this effect appears to be negligible. The resistance of the molecule–substrate junction is much smaller than the impedance of the vacuum gap due to a sufficient electron density of states at the ethynyl spacer (figure 2(b)).
- 5
More precisely, the force F was calculated from the measured shift of the quartz sensor resonance (supplementary material, section 2, available at stacks.iop.org/njp/17/013012/mmedia).
- 6
Figures 1(c) and (d) suggest that Zn-porphyrin may start to rotate around the etynyl spacer induced by the tunneling electrons. This rotation may influence the orbitals localized at the ethynyl spacer and thus the conductance. Owing to the time resolution in the measurements (100 ms per point) no conductance variations are observed.