


Abstract
We show by employing density-functional theory calculations (including a hybrid functional) that ZnO surfaces can be stabilized by bulk dopants. As an example, we study the bulk-terminated ZnO ( ) surface covered with half a monolayer of hydrogen. We demonstrate that deviations from this half-monolayer coverage can be stabilized by electrons or holes from bulk dopants. The electron chemical potential therefore becomes a crucial parameter that cannot be neglected in semiconductor surface studies. As one result, we find that to form the defect-free surface with a half-monolayer coverage of hydrogen for n-type ZnO, ambient hydrogen background pressures are more conducive than high vacuum pressures.
) surface covered with half a monolayer of hydrogen. We demonstrate that deviations from this half-monolayer coverage can be stabilized by electrons or holes from bulk dopants. The electron chemical potential therefore becomes a crucial parameter that cannot be neglected in semiconductor surface studies. As one result, we find that to form the defect-free surface with a half-monolayer coverage of hydrogen for n-type ZnO, ambient hydrogen background pressures are more conducive than high vacuum pressures.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Density-functional theory (DFT) calculations in combination with ab initio thermodynamics have contributed significantly to the development of an atomistic understanding of surface structures in material and surface science [1–4]. In this approach, externally controlled factors, such as temperature or partial pressures of the surrounding environment, are considered through the atomic chemical potentials. However, the role of the electron chemical potential for surfaces has mostly been ignored. Its role has so far only been investigated for defects in the bulk or at interfaces [5–8]. Here we build on these studies and show that free charge carriers from bulk dopants can stabilize surface structures that exhibit partially occupied dangling bonds at the surface. We demonstrate that it is crucial to not only consider the atomic chemical potentials in first-principles surface studies but also to take the electron chemical potential into account.
The structures of the polar ZnO surfaces are still debated fervidly in the literature [9–14]. Experimentally, the exact surface structures for ZnO ( ) could so far not be identified unambiguously [9, 10]. A structure that has been proposed is a fractional monolayer of disordered H atoms adsorbed on O sites [10, 11, 15, 16]. We will study this structure in more detail and show that the hydrogen concentration at the surface depends on the electron chemical potential (Fermi energy). We find that the stability region of the semiconducting half-monolayer structure is considerably decreased in the surface phase diagram when taking the contribution of bulk dopants into consideration. Therefore, for n-type materials with high donor concentrations (e.g. ZnO), for which the electron chemical potential is close to the conduction-band minimum (CBm), the effect of the electron chemical potential deserves particular attention. First experimental evidence for the role of the electron chemical potential for semiconductor surfaces has been reported for strongly doped GaAs [17]. Our findings should also apply to other surfaces of ZnO and other semiconductors.
) could so far not be identified unambiguously [9, 10]. A structure that has been proposed is a fractional monolayer of disordered H atoms adsorbed on O sites [10, 11, 15, 16]. We will study this structure in more detail and show that the hydrogen concentration at the surface depends on the electron chemical potential (Fermi energy). We find that the stability region of the semiconducting half-monolayer structure is considerably decreased in the surface phase diagram when taking the contribution of bulk dopants into consideration. Therefore, for n-type materials with high donor concentrations (e.g. ZnO), for which the electron chemical potential is close to the conduction-band minimum (CBm), the effect of the electron chemical potential deserves particular attention. First experimental evidence for the role of the electron chemical potential for semiconductor surfaces has been reported for strongly doped GaAs [17]. Our findings should also apply to other surfaces of ZnO and other semiconductors.
2. Charged surfaces in the supercell method
In figure 1 the ZnO ( ) surface with half a monolayer of H atoms is shown, which is denoted by 2 × 1-H in the following. The bulk truncated (
) surface with half a monolayer of H atoms is shown, which is denoted by 2 × 1-H in the following. The bulk truncated ( ) surface is terminated with O atoms, each of which is bonded to the three Zn atoms below. This leads to 1.5 electrons in the dangling bonds of the O atoms. By transferring 0.5 electrons from one O atom to every second O atom, one fully occupied lone pair is formed. The other O atom with one electron remaining is saturated by forming a bond with an H atom. This occurs, for example, in organometallic vapor-phase epitaxy, for which excess H atoms are available from the residual gas [9, 10]. DFT calculations employing the Perdew–Burke–Ernzerhof (PBE) [18] functional find that this H-stabilized bulk-terminated surface has the lowest formation energy for all experimentally reachable H pressures at 800 K [15]. Removing a single H atom would produce a dangling bond occupied with one electron, which can be saturated by one excess electron. This electron could be provided by the bulk and would then form a lone pair.
) surface is terminated with O atoms, each of which is bonded to the three Zn atoms below. This leads to 1.5 electrons in the dangling bonds of the O atoms. By transferring 0.5 electrons from one O atom to every second O atom, one fully occupied lone pair is formed. The other O atom with one electron remaining is saturated by forming a bond with an H atom. This occurs, for example, in organometallic vapor-phase epitaxy, for which excess H atoms are available from the residual gas [9, 10]. DFT calculations employing the Perdew–Burke–Ernzerhof (PBE) [18] functional find that this H-stabilized bulk-terminated surface has the lowest formation energy for all experimentally reachable H pressures at 800 K [15]. Removing a single H atom would produce a dangling bond occupied with one electron, which can be saturated by one excess electron. This electron could be provided by the bulk and would then form a lone pair.
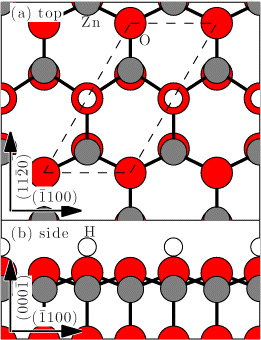
Figure 1. (a) Top view of the ( ) ZnO surface (Zn atoms are gray and O atoms red) with half a monolayer H (white). This structure is denoted 2 × 1-H. The 2 × 1 unit cell is indicated by a dashed line. (b) Side view of the (
) ZnO surface (Zn atoms are gray and O atoms red) with half a monolayer H (white). This structure is denoted 2 × 1-H. The 2 × 1 unit cell is indicated by a dashed line. (b) Side view of the ( ) ZnO surface.
) ZnO surface.
Download figure:
Standard image High-resolution imageTo study charged surfaces which deviate from the half-monolayer H coverage, we have to introduce a reservoir for the excess charge to make surfaces with different amounts of excess charge comparable with each other. This is analogous to bulk defect calculations. To determine the lowest-energy state, we compare the relative energies of the different charge states. To alter the charge state, electrons must be taken from or removed to a reservoir. The energy of the electrons is determined by the electron chemical potential μe and the formation energies therefore depend on the position of it. The formation energy ΔHf of a H-covered ZnO( ) surface with an excess charge q is given by the difference of the total energy of the slab system, Eslabq, the total energy of the neutral bulk, Ebulk0, the chemical potential of H, μH, the number of H atoms, NH and the charging energy qμe:
) surface with an excess charge q is given by the difference of the total energy of the slab system, Eslabq, the total energy of the neutral bulk, Ebulk0, the chemical potential of H, μH, the number of H atoms, NH and the charging energy qμe:

In this paper, all slabs have the same number of Zn and O atoms, and only the chemical potential of H appears.
For a slab system in a supercell an excess charge cannot be created in the same way as for a bulk system where the charges are compensated by a uniform background of opposite charge [19]. This uniform background leads to a diverging energy with increasing vacuum between the slabs, as illustrated in figure 2. Recently, an auxiliary field to compensate this dipole was introduced [20]. Here we follow a different strategy. We confine the compensating background to have the same spatial extent as the extra holes or electrons. We could again distribute the compensating charge homogeneously within these confines, but in our all-electron code it is easier to introduce point charges at atomic sites. Both the nuclear and electronic charge of each O atom are slightly modified. The excess charge goes to the valence or conduction band, simulating free charge carriers, whereas the modified nuclear charge simulates the charged background. How one avoids spurious total energy contributions from these altered nuclei will be addressed below. Overall the supercell remains neutral and the problem of a diverging energy with increasing vacuum separation between the slabs is solved. This approach is commonly called the virtual crystal approximation (VCA) and has previously been used to calculate the formation energies of charged surfaces in the supercell method [21, 22]. The VCA enables the calculation of charged surfaces exactly in the same manner as for charged bulk defects [8, 23–25].
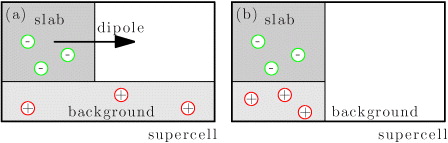
Figure 2. (a) The introduction of negative charge into a periodical supercell requires a positive compensating background. The uniform background leads to a dipole, which diverges upon increasing the amount of vacuum in the supercell. (b) Confining the compensating charge to the extension of the slab in the surface supercell prevents the build up of an undesired dipole.
Download figure:
Standard image High-resolution imageThe electron chemical potential μe in equation (1) is a variable in our formalism that for non-degenerate semiconductors can vary from the valence band maximum (VBM) to the CBm. Our interest in this work is the effect of e.g. one excess charge carrier at the surface and whether it is energetically favorable to saturate the dangling bond or to adsorb a hydrogen atom instead. The excess charge in the supercell must therefore not be confused with an effective doping concentration. For a finite amount of excess charge q and an increasing size of the supercell, the effective carrier concentration in the calculations goes to zero. In our case, the charge addition is governed by a reservoir, which is given by the charging energy qμe. We have thus effectively decoupled the electron chemical potential from the excess charge.
In the VCA, the total energies of the charged slab Eslabq and the neutral bulk Ebulk0 cannot be subtracted from each other directly, because of their different nuclear charges. We therefore have to eliminate Ebulk0 from equation (1). To do so, we exploit the fact that the total energies of the neutral and the charged bulk Ebulkq are related by the electron chemical potential of the charged bulk:

where N is the number of electrons. This approximation, which holds for small charges q, is warranted in our calculations, because q never exceeds 1.6 × 10−2 per ZnO pair. Inserting equation (2) into equation (1) the formation energy becomes
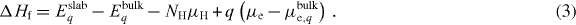
The VBM and CBm shift by approximately ±0.03 eV when decreasing or increasing the nuclear charge by 1.6 × 10−2 per ZnO pair. Therefore, we define the chemical potential relative to the O2p band, the VBM of the bulk in the uncharged cell  VBM'q:
VBM'q:

The relative electron chemical potential Δμe has values between zero and the band gap. Analogously, a relative hydrogen chemical potential ΔμH is introduced, which is zero at half of the total energy of the H2 molecule μH2. The formation energy is then given by:
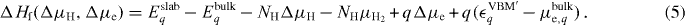
We have now expressed the formation energy entirely in quantities of the charged systems with the same q. Therefore, other contributions, such as the electrostatic energy of the core electrons arising from the fractional change of the nuclear charges in the VCA, compensate each other.
As alluded to before, equation (5) embodies the essence of our approach, which permits us to introduce the electron chemical potential of the bulk as an independent variable in our calculations and to decouple it from the excess charge q in the valence or conduction band. Our approach does not explicitly take band bending, which is a result of charge transfer from the bulk to the surface, into account for arbitrary electron chemical potentials. However, in our calculations the number of excess charge carriers is approximately 1 × 1021 cm−3. Therefore, the associated depletion or accumulation layer is always equal or smaller than the slab thickness and the effects of band bending are automatically included in the calculations. To analyze the contribution of band bending to the total energy, we used a simple electrostatic capacitor model [26] and verified that it is smaller than 0.06 eV/(6 × 6). This is in agreement with tests conducted for varying the excess charge carrier concentration numerically by changing the supercell size. For low carrier concentrations, the energy contribution from band bending will become more significant. We expect this to stabilize more ordered structures. However, for these concentrations, the band bending contributions are no longer reliably described by our slab approach. Appropriate corrections would have to be added for which we refer to future work. Here, we will focus on electron chemical potentials close to the conduction band in the limit of large doping concentrations.
3. Computational details
The formation energies are calculated using equation (5) and DFT [27] as implemented in the code FHI-aims3 [28]. The surface supercell was chosen to be 6 × 6, with the slab containing four ZnO double layers, and the bottom of the slab saturated by pseudo-H atoms. We used a 3 × 3 k-point mesh. The layer of H atoms as well as the top three layers of the slab were relaxed until the forces were smaller than 10−3 eV Å−1. The PBE functional [18] and the Heyd–Scuseria–Ernzerhof functional [29] were used. The parameter that controls the screening length was set to 0.2 Å−1 for both PBE and Hartree–Fock exchange. The mixing parameter that controls the amount of exact exchange was chosen to be 0.4 (very close to the 0.375 of [30]) so that both the calculated valence bandwidth [31] and the band gap of bulk ZnO match their experimental values within an error of only 0.1 eV.
4. Charged H-terminated ZnO surfaces
The formation energies calculated with PBE for the 2 × 1-H surface are shown in figure 3(a) for different q as a function of the electron chemical potential Δμe. We use the formation energy of the neutral surface (q = 0.0) as reference and set it to 0 eV. The range of the electron chemical potential Δμe is equal to the band gap and was determined from bulk calculations. It can vary between p-type (electron-poor) conditions Δμe = 0.0 eV and n-type (electron-rich) conditions Δμe = 0.76 eV. The much too small band gap (3.3 eV in the experiment [32]) is largely an effect of the self-interaction error inherent in the PBE functional and the absence of the derivative discontinuity. Later we will also show results for the hybrid functional [29], where the band gap compares much better with experiment. In figure 3(a) it can be seen that for almost all electron chemical potentials the neutral charge state q = 0.0 is lowest in energy. For the semiconducting 2 × 1-H surface it is difficult to add or remove an electron because it would cost as much as the band gap.
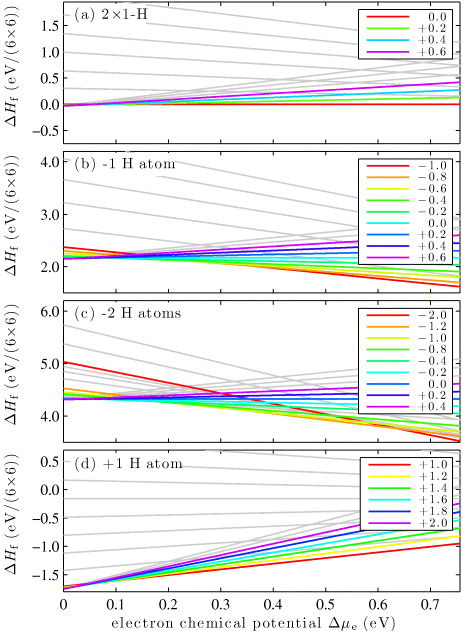
Figure 3. The formation energy ΔHf as a function of the electron chemical potential Δμe of the ( ) ZnO surface for different H coverages and charge states. Only those charge states are labeled that are stable within the range of the electron chemical potential. All others are shown in light gray. (a) The 2 × 1-H surface, (b) one H atom missing, (c) two H atoms missing from the 2 × 1-H and (d) one H atom added to the 2 × 1-H.
) ZnO surface for different H coverages and charge states. Only those charge states are labeled that are stable within the range of the electron chemical potential. All others are shown in light gray. (a) The 2 × 1-H surface, (b) one H atom missing, (c) two H atoms missing from the 2 × 1-H and (d) one H atom added to the 2 × 1-H.
Download figure:
Standard image High-resolution imageIn the next step, we remove a single H atom from a 6 × 6 unit cell of the 2 × 1-H surface, which leaves a single dangling bond occupied with one electron. The formation energies for different charge states are shown in figure 3(b). For n-type conditions (Δμe > 0.4 eV), the surface with one extra electron (q = −1.0) is lowest in energy. This means that it is energetically favorable to transfer one electron from the bulk to the dangling bond with one single electron. For other conditions (Δμe < 0.4 eV), a less negatively or even positively charged surfaces become stable.
The results of removing two H atoms from a 6 × 6 unit cell can be seen in figure 3(c). For n-type conditions the surface with two extra electrons (q = −2.0) is lowest in energy. Two fully occupied lone pairs are formed. Further calculations showed that the two newly formed lone pairs repel each other. The energy cost to move the lone pairs next to each other from their furthest apart configuration is approximately 0.1 eV. Removing three or more H atoms would require excess charges that are too large for our scheme, and was therefore not considered.
We can also adsorb one additional H atom on the 2 × 1-H surface. For this, an electron has to be removed from a lone pair to form a dangling bond with a single electron. At this site, the H atom can then be adsorbed. This is reflected by the observation that in figure 3(d) the surface with one hole (q = + 1.0) is the lowest in energy for almost the entire range of electron chemical potentials.
5. Comparing surfaces with different numbers of H atoms
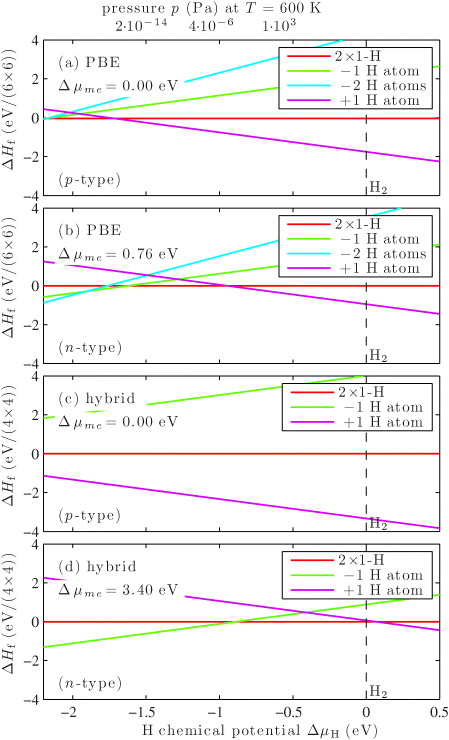
To compare surface structures with different numbers of H atoms, we plot in figure 4 the formation energy as function of the H chemical potential ΔμH for p- and n-type conditions and for the PBE and the hybrid functional. At a given electron chemical potential the surfaces structures with the lowest formation energy were determined from figure 3. For T = 0 K and ΔμH = 0 eV, the surface is in equilibrium with an H2 gas. Using an ideal-gas-like reservoir for H2, the H chemical potential ΔμH can be written as a function of the temperature T and the pressure p [2, 3, 6]. The resulting pressure for a temperature of 600 K, which is a typical growth and annealing temperature, is given on the top axis. Temperature effects due to vibrations will affect the formation energies [13]. However, as all surfaces considered here have very similar geometries, temperature effects will not change the conclusions of this paper.
{kind=link}
{kind=link}
{kind=link}
Figure 4. The four surface structures considered are plotted as a function of the H chemical potential ΔμH for (a) PBE functional and p-type conditions, (b) PBE functional and n-type conditions, (c) hybrid functional and p-type conditions and (d) hybrid functional and n-type conditions.
Download figure:
Standard image High-resolution image{kind=link}
First we discuss the PBE results in figures 4(a) and (b). In figure 4(a) the formation energies for p-type conditions are shown for four different H coverages. Although until now it is not possible to grow p-type bulk crystals [32], we nonetheless show p-type conditions for completeness and to compare them with the n-type crystals later. The 2 × 1-H surface is stable only below ΔμH = −1.7 eV and above an additional H atom can be stabilized. Experimentally, ZnO crystals exhibit n-type behavior [32], which is shown in figure 4(b). The 2 × 1-H surface is only stable for H chemical potentials from −1.6 to −1.0 eV. For smaller ΔμH, missing H atoms, and for larger H chemical potential, additional H atoms would be stable. However, these results are based on PBE calculations, which are plagued by two deficiencies. Firstly, the calculated band gap of ZnO is only 0.76 eV compared to 3.3 eV in experiment [32]. Secondly, the O2 and OH binding energies are more consistently described using exchange-correlation functionals including a fraction of exact exchange as can be seen in table 1.
Table 1. Atomization energies of H2, O2 and OH for the PBE and hybrid functional compared with highly accurate quantum-chemistry W4 calculations [33].
| H2 (eV) | O2 (eV) | OH (eV) | |
|---|---|---|---|
| PBE | 4.54 | 6.23 | 4.76 |
| Hybrid | 4.52 | 4.89 | 4.47 |
| W4 | 4.75 | 5.24 | 4.65 |
To remedy both deficiencies, we perform calculations using a hybrid functional [29]. Owing to the larger computational expense, the calculations were restricted to 4 × 4 surface unit cells. For this reason, we omitted the structure with two missing H atoms. Because both the OH molecular bond length and the ZnO lattice constant are reduced by 2% in the hybrid calculations, the relaxed geometries were taken from the PBE calculations and scaled down by 2%.
The band gap of 3.40 eV calculated with the hybrid functional is only slightly larger than the experimental one of 3.3 eV [32]. The effect of the larger theoretical band gap can be seen in figure 4(c). In contrast to PBE (figure 4(a)), the surface with one additional H atom is stable for all H chemical potentials. In the hybrid calculations, this surface is more than 2 eV lower in energy, which is mostly a result of the larger range of electron chemical potentials due to the larger and more realistic band gap from the hybrid calculations. The formation energies of charged surfaces, therefore vary much stronger with the electron chemical potential. Based on our hybrid calculations, we conclude that for p-type conditions H-rich surfaces should be observed experimentally for all pressures. This is a direct consequence of the larger OH binding energy, which leads to very-low surface formation energies.
With our approach, we cannot predict larger modifications of the H coverage. However, in experiment, both the full H coverage or the full H removal should be energetically unfavorable owing to the energy contribution of band bending arising from the substantial charge transfer from the bulk to the surface.
The result of the second PBE deficiency, namely, inaccurate O2 and OH binding energies, can be seen in figure 4(d), where the formation energies for n-type conditions in the hybrid functional are shown. Compared to the PBE energies in figure 4(b) the energies are shifted relative to each other, because they have different numbers of O lone pairs and OH bonds. For the hybrid functional the structure with one missing H atom is stable for H chemical potential smaller than −0.9 eV (10−4 Pa at 600 K). For larger H chemical potential the 2 × 1-H surface becomes stable, whereas the structure with an additional H atom is never stable. This suggests that in experiment the 2 × 1-H surface should be favored for an ambient H2 gas background pressure. Using such conditions should lead to better defined surfaces, which could be advantageous for a more controlled formation of interfaces with other materials. All things considered for hybrid functional calculations the stability windows are shifted to larger H chemical potential. Therefore, to obtain the correct pressure and temperature dependence, calculations with a semi-local exchange-correlation functional do not suffice, but calculations with a hybrid functional are imperative.
6. Summary and conclusion
In summary, surfaces can be stabilized by electrons or holes from bulk dopants. As a consequence, deviations from the half-monolayer H coverage can be stable for ZnO ( ) surface depending on the H conditions and the electron chemical potential. Furthermore, for ZnO high negative charge concentrations at the surface have been observed [34], confirming our hypothesis. The triangular reconstructions observed [12] for the Zn-terminated side, which have partially occupied dangling bonds, could also be stabilized through bulk defects, most likely in connection with a mechanism proposed in [35]. Based on these observations, we conclude that free carriers can stabilize surface structures that might have previously been discounted. For semiconductors with high carrier concentrations, it is crucial that the electron chemical potential is taken into consideration when exploring surface structures.
) surface depending on the H conditions and the electron chemical potential. Furthermore, for ZnO high negative charge concentrations at the surface have been observed [34], confirming our hypothesis. The triangular reconstructions observed [12] for the Zn-terminated side, which have partially occupied dangling bonds, could also be stabilized through bulk defects, most likely in connection with a mechanism proposed in [35]. Based on these observations, we conclude that free carriers can stabilize surface structures that might have previously been discounted. For semiconductors with high carrier concentrations, it is crucial that the electron chemical potential is taken into consideration when exploring surface structures.
Acknowledgments
We thank N A Richter, S V Levchenko, S Blumstengel and F Henneberger for valuable comments and the DFG collaborative research project 951 'HIOS' for financial support. YX acknowledges support from the Alexander von Humboldt foundation.
Footnotes
- 3
Fritz Haber Institute ab initio molecular simulations package.