


Abstract
The adsorption of benzene on metal surfaces is an important benchmark system for hybrid inorganic/organic interfaces. The reliable determination of the interface geometry and binding energy presents a significant challenge for both theory and experiment. Using the Perdew–Burke–Ernzerhof (PBE), PBE + vdW (van der Waals) and the recently developed PBE + vdWsurf (density-functional theory with vdW interactions that include the collective electronic response of the substrate) methods, we calculated the structures and energetics for benzene on transition-metal surfaces: Cu, Ag, Au, Pd, Pt, Rh and Ir. Our calculations demonstrate that vdW interactions increase the binding energy by more than 0.70 eV for physisorbed systems (Cu, Ag and Au) and by an even larger amount for strongly bound systems (Pd, Pt, Rh and Ir). The collective response of the substrate electrons captured via the vdWsurf method plays a significant role for most substrates, shortening the equilibrium distance by 0.25 Å for Cu and decreasing the binding energy by 0.27 eV for Rh. The reliability of our results is assessed by comparison with calculations using the random-phase approximation including renormalized single excitations, and the experimental data from temperature-programmed desorption, microcalorimetry measurements and low-energy electron diffraction.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Hybrid inorganic/organic systems (HIOS) have attracted considerable interest because they can be used to tailor the electronic, optical and magnetic properties of functional materials [1–5]. A wide range of HIOS have been created through the self-assembly of organic molecules on diverse substrates [6–8]. Special attention has been paid to the adsorption of benzene (Bz) on transition-metal surfaces [9–30]. Bz is a fundamental aromatic molecule and, more importantly, the prototype for an entire class of aromatic hydrocarbons [30–33]. Such aromatic hydrocarbons are among the principal constituents of biochemical and petroleum industries, and they are widely used in the production of plastics, fibers and rubbers. Various useful aromatic molecules are produced from Bz by catalysis, including bromobenzene, alkylbenzene and nitrobenzene [34, 35].
Despite considerable progress in experimental techniques, information obtained from experiments on the adsorbate geometry, adsorption energy and electronic properties of HIOS is often hard to interpret, or even lacking. For example, due to the relative difficulty of controlling and measuring weakly bound systems, no experimental adsorption height has been reported so far for Bz physisorbed on noble metals. The only experimentally deduced adsorption height, to the best of our knowledge, was determined for the disordered Bz chemisorbed on the Pt(111) surface at a coverage close to or less than one [36].
The binding energy, which reflects the strength of the interaction between an adsorbate and the substrate, is another key parameter for the description of HIOS. Experimental binding energies are mainly obtained by temperature-programmed desorption (TPD) [22–25, 27] and microcalorimetry measurements [32, 37–39]. TPD is the most extensively used method for determining the kinetic and thermodynamic parameters of desorption processes and decomposition reactions. The desorbing molecular species are selected by their mass, while the amount of adsorbate is determined by integrating the peaks of the desorption spectrum. The Redhead formula is typically used to calculate the adsorption energy based on three parameters: the desorption temperature, the heating rate and a pre-exponential factor [40]. The wide range of empirical pre-exponential factors (1013–1019 s−1) that are typically used for molecular desorption may cause a notable uncertainty in the determined binding energy [41–43]. TPD experiments have been carried out to study the interaction of Bz with the Cu [13], Ag [24] and Au surfaces [29]. However, special attention must be paid to the interpretation of TPD spectra for the Pd, Pt and Rh surfaces, because the adsorbed Bz molecules may decompose during heating, in particular at low coverage [44]. Here, we revisit the adsorption energies from the measured TPD spectra for Bz on Cu(111), Ag(111) and Au(111) using the recently published Campbell–Sellers method [43].
In contrast to TPD, microcalorimetry measurements directly determine the heat released during the adsorption process [32, 37–39]. In principle, calorimetry measurements are applicable to many adsorption systems, no matter whether the adsorbed molecules dissociate, decompose or react with other species [39]. King's group first demonstrated that calorimetry is a valuable method for precise measurements of heats of adsorption for small molecules (CO, O2, NO, C2H2, C2H4, CO/K and O2/K) on single-crystal surfaces (Ni, Pd, Pt and Rh) [39]. Utilizing a removable pyroelectric heat detector, Campbell's group developed a calorimetry instrument that allows measurement of heats of adsorption for larger organic molecules, e.g. Bz and naphthalene, on the Pt(111) surface [32, 37, 38, 45].
The adsorption of Bz on metals has also been intensively studied theoretically. First-principles calculations were predominantly carried out using density-functional theory (DFT) with various flavors of the generalized gradient approximation (GGA) for the exchange–correlation functional. The GGA-PBE (Perdew–Burke–Ernzerhof) functional sometimes yields seemingly accurate results for the chemisorption of small molecules, but this is not always the case, as exemplified by the 'CO adsorption puzzle' [46, 47]. When van der Waals (vdW) interactions play a crucial role in adsorption processes, further problems appear since commonly used local density [48], gradient-corrected [49–51] and hybrid functionals [52–54] are unable to capture the long-range vdW tails. This leads to a substantial overestimation of adsorption heights and an underestimation of binding energies [20, 55]. In the last decade, several vdW-inclusive approaches in the DFT framework have been developed and employed to study the interaction between Bz and coinage metal surfaces [10, 16, 17, 20, 28, 56–59]. Accounting for vdW interactions in the standard DFT functionals brings a large increase in binding energies, and results in a much better agreement with experiments. The strongly bound, chemisorbed systems Bz/Pd(111) [60–62], Bz/Pt(111) [60–67] and Bz/Rh(111) [62] have been studied with the GGA functionals PW91, PBE and BP86. In these systems, covalent bonding is often recognized as the dominant interaction, assuming that vdW effects will only play a minor role. However, as exemplified in [32] and in our current work for the Bz/Pt(111) system, the PBE and PW91 adsorption energies (0.85–1.21 eV) [60–66] only account for half of the adsorption energy determined from microcalorimetry measurements (1.57–1.91 eV) [32, 37].
The role of vdW interactions in the binding between small molecules in the gas phase is well understood and properly described by the methods of quantum chemistry. In the DFT framework, recently, several promising vdW-inclusive approaches, such as DFT-D3 [28], vdW-DF2 [68], vdW-DF-type functionals with modified exchange [69], the DFT + XDM method [70, 71] and the DFT + vdW method [72, 73], have shown remarkable accuracy for intermolecular interactions (see [74] for a concise review of vdW-inclusive methods in DFT). However, none of the mentioned methods accurately account for non-local (inhomogeneous) collective electron response in the vdW energy tail, an effect that is particularly important in metals [75, 76]. Recently, some of the authors have developed a method to calculate the vdW energy for atoms and molecules on surfaces by using a combination of the DFT + vdW approach for intermolecular interactions with the Lifshitz–Zaremba–Kohn (LZK) theory [75] for the non-local collective response within the substrate surface. This method will be referred to as 'DFT + vdWsurf' in the following. Calculations using the DFT + vdWsurf method have demonstrated that the inclusion of these collective effects, which go beyond the atom-based pairwise description of vdW interactions, enables us to reliably describe the binding in many systems, including the Xe atom, benzene, 2-pyrrolidone, naphthalene and 3,4,9,10-perylene-tetracarboxylic acid dianhydride adsorbed on several close-packed transition-metal surfaces [58, 76–79].
In this paper, we carry out a systematic study of Bz adsorbed on transition-metal surfaces by means of the PBE, PBE + vdW [72] and PBE + vdWsurf methods [76]. The accuracy of our results is assessed by comparison with calculations using the random-phase approximation plus renormalized single excitations (the EX + cRPA + rSE approach [80]). The RPA method represents the fifth (and highest) rung of 'Jacob's ladder' of Perdew [81], and seamlessly includes vdW interactions and collective screening effects by the metal electrons. The rSE correction alleviates the underbinding problem of RPA, and adding rSE to RPA yields accurate binding energies [80]. In the remainder of this paper, we will present a brief introduction to the DFT + vdWsurf method (section 2). Section 3 deals with the lattice constants of bulk metals from different approaches. In section 4, we provide a detailed description of the adsorption models and DFT calculation parameters. The binding energies obtained from EX + cRPA calculations for Bz on the coinage metal clusters are presented in section 5. In section 6, we will explain our procedure to calculate the adsorption energies from TPD measurements. From sections 7 to 9 we will discuss the potential-energy surface (PES), the adsorption sites and the role of vdW interactions and collective screening effects on the binding structures and energetics in the weakly bound (Cu, Ag and Au) and strongly bound (Pd, Pt, Rh and Ir) systems.
2. Theoretical methods
2.1. Density-functional theory (DFT) calculations
The DFT calculations presented in this paper employed the numeric atom-centered basis set all-electron code FHI-aims [82, 83], together with the PBE exchange–correlation functional [50]. We employ the vdW-inclusive PBE + vdW [72] and PBE + vdWsurf approaches [76] to account for vdW interactions and collective response effects. The scaled zeroth-order regular approximation [84] was applied for treating relativistic effects. The 'tight' settings including the 'tier2' standard basis set in the FHI-aims code (Fritz Haber Institute ab initio molecular simulations) were used for H and C, and 'tier1' for transition metals. A convergence criterion of 10−2 eV Å−1 for the maximum final force was used for structural relaxations. Also convergence criteria of 10−5 electrons per unit volume for the charge density and 10−4 eV for the total energy of the system were utilized for all computations. Adopting these settings, the numerical accuracies in determining the binding energy and equilibrium distance are better than 0.01 eV and 0.01 Å, respectively.
2.2. DFT + vdWsurf method
The DFT + vdWsurf method consists of an extension to standard interatomic vdW approximations for the modeling of adsorbates on surfaces [76]. This is achieved by combining the DFT + vdW method with the LZK theory for the vdW interaction between an atom and a solid surface [75, 85]. Here we use the PBE functional and we refer to this approach as PBE + vdWsurf throughout the paper. In analogy to the PBE + vdW method, the PBE + vdWsurf approach adds the dispersion energy to the DFT total energy given as a sum of −Cab6R−6ab terms, where Rab is the distance between atoms a and b. We include, however, the collective many-body response (screening) of the substrate electrons, captured via the vdWsurf method, in the determination of the C6 coefficients and vdW radii (vdW parameters), going effectively beyond the interatomic pairwise description. This is achieved through the dependence of the vdW parameters on the dielectric function of the substrate [75, 86]. The frequency-dependent dielectric functions for transition metals,  (iω), were taken from reflection electron energy-loss spectroscopy (REELS) experiments [87]. The only adjustable parameter is the cut-off sR in the damping function of the vdW tail at close interatomic distances. This is kept the same as in the PBE + vdW method, which is rationalized by the fact that the cohesive properties of solids are not sensitive to a reasonable variation of sR when using the screened PBE + vdW method [88]. Table 1 shows the effect that the collective response of the metal electrons has on the vdW parameters for different bulk metals, reducing the vdW C6 coefficients up to a factor of five with respect to their free atom counterparts in coinage metals. We note in passing that the PBE + vdWsurf method also includes image-plane effects, owing to the interatomic projection. The LZK theory is exact for atom–surface distances beyond orbital overlap, but it does not include effects due to rapid spatial variations (interface polarization) in the dielectric function close to the surface. However, by linking the LZK theory to the PBE + vdW method these aspects are automatically included in our calculations. Specifically, this is achieved through the dependence of the screened Cab6 interatomic coefficients on the self-consistent electron density via Hirshfeld partitioning [89, 90]. In fact, we have found that the C6 coefficient for the Pt atoms in the first layer is 3.5% smaller relative to the metal atoms inside the bulk. Also the C6 value for the Bz molecule increases by 26.5% upon adsorption on Pt(111), with respect to the free Bz molecule. It has already been shown that the DFT + vdWsurf method yields excellent agreement with experiments for a variety of atoms and molecules adsorbed on metal surfaces [58, 76–79]. We remark that the DFT + vdWsurf method can be applied in general to insulator, semiconductor and metal surfaces. Currently, its performance is also being assessed for non-close-packed surfaces and surfaces with defects.
(iω), were taken from reflection electron energy-loss spectroscopy (REELS) experiments [87]. The only adjustable parameter is the cut-off sR in the damping function of the vdW tail at close interatomic distances. This is kept the same as in the PBE + vdW method, which is rationalized by the fact that the cohesive properties of solids are not sensitive to a reasonable variation of sR when using the screened PBE + vdW method [88]. Table 1 shows the effect that the collective response of the metal electrons has on the vdW parameters for different bulk metals, reducing the vdW C6 coefficients up to a factor of five with respect to their free atom counterparts in coinage metals. We note in passing that the PBE + vdWsurf method also includes image-plane effects, owing to the interatomic projection. The LZK theory is exact for atom–surface distances beyond orbital overlap, but it does not include effects due to rapid spatial variations (interface polarization) in the dielectric function close to the surface. However, by linking the LZK theory to the PBE + vdW method these aspects are automatically included in our calculations. Specifically, this is achieved through the dependence of the screened Cab6 interatomic coefficients on the self-consistent electron density via Hirshfeld partitioning [89, 90]. In fact, we have found that the C6 coefficient for the Pt atoms in the first layer is 3.5% smaller relative to the metal atoms inside the bulk. Also the C6 value for the Bz molecule increases by 26.5% upon adsorption on Pt(111), with respect to the free Bz molecule. It has already been shown that the DFT + vdWsurf method yields excellent agreement with experiments for a variety of atoms and molecules adsorbed on metal surfaces [58, 76–79]. We remark that the DFT + vdWsurf method can be applied in general to insulator, semiconductor and metal surfaces. Currently, its performance is also being assessed for non-close-packed surfaces and surfaces with defects.
Table 1. Screened vdW parameters as used in the PBE + vdWsurf method. For comparison, free atom parameters as used in the PBE + vdW method are also shown. C6 (in hartree bohr6), R0 (in bohr) and α (in bohr3) denote the dispersion coefficient, vdW radius and polarizability, respectively.
| Substrate | Free atom | Screened | ||||
|---|---|---|---|---|---|---|
| C6 | R0 | α | C6 | R0 | α | |
| Cu | 253 | 3.76 | 42.0 | 59 | 2.40 | 10.9 |
| Ag | 339 | 3.82 | 50.6 | 122 | 2.57 | 15.4 |
| Au | 298 | 3.86 | 36.5 | 134 | 2.91 | 15.6 |
| Pd | 158 | 3.66 | 23.7 | 102 | 3.07 | 13.9 |
| Pt | 347 | 3.92 | 39.7 | 120 | 2.80 | 14.5 |
| Rh | 469 | 3.95 | 56.1 | 84 | 2.42 | 13.0 |
| Ir | 359 | 4.00 | 42.5 | 98 | 2.71 | 13.2 |
3. Metal bulk lattice constants
We have calculated the lattice constants of the bulk metals by fitting the Birch–Murnaghan equation of state to DFT cohesive-energy curves [91]. The vdW energy was added to the PBE functional using the PBE + vdW [72] and PBE + vdWsurf [76] approaches, and for the sampling of the Brillouin zone we employed a 16 × 16 × 16 Monkhorst–Pack mesh for the primitive cell. Table 2 shows that the standard PBE functional underbinds, and leads to an overestimation of the lattice constants for all the studied metals. Upon inclusion of the vdW energy via the PBE + vdW approach, the lattice constants are reduced, and as a result are in better agreement with the experimental data, except for Cu. The improvement obtained with PBE + vdW is surprising if we consider that screening of the vdW energy between ions embedded in a metallic electron background is known to be significant [92]. These effects are not included in the PBE + vdW approach. The observed improvement of the metallic bulk lattice constants arises due to two opposing contributions: (i) overestimation of the long-range vdW energy, due to the use of large free-atom metallic C6 coefficients (see table 1) and, (ii) large values of vdW radii, which partially compensate the overestimation of the long-range vdW energy. In contrast, the PBE + vdWsurf method simulates to some extent the screening by the metallic bulk electrons. However, for the vdW energy inside the metallic bulk, the PBE + vdWsurf method overestimates the interaction between metallic electrons, which is already described accurately by the PBE functional (since PBE reduces to LDA for homogeneous electron densities). This effect does not pose a problem for molecules adsorbed on surfaces, because the adsorbate interacts with both the localized ions and the delocalized metallic electrons. Unlike the PBE + vdW method, the PBE + vdWsurf approach does not result in an artificial cancelation between the short- and long-range vdW energies. This leads sometimes to a slight increase of the lattice constants compared to the PBE functional (for Au, Pd, Pt and Ir metals), while decreasing the lattice constant for all the other cases. Overall, the performance of the PBE + vdWsurf method for metal lattice constants is as good as or better than PBE. Further improvement of the lattice constants requires a full microscopic treatment of the polarizability due to localized ions and metallic electrons. Note that the vdW-DF functional systematically overestimates the lattice constants presumably due to the revPBE exchange functional [93]. Some of the empirically optimized exchange functionals, such as optB86b [69, 93] and C09 [94, 95], are found to reduce the separations.
Table 2. Lattice constants (in Å) of bulk metals used in this work, which are determined from the PBE, PBE + vdW and PBE + vdWsurf methods. Experimental lattice constants, which are extrapolated to 0 K and corrected with the zero-point anharmonic expansion, are listed for comparison [96]. The experimental values in parentheses include the contribution from the zero-point vibrations.
| Metal | PBE+vdWsurf | PBE+vdW | PBE | Experiment |
|---|---|---|---|---|
| Cu | 3.572 | 3.543 | 3.631 | 3.596 (3.603) |
| Ag | 4.007 | 4.071 | 4.149 | 4.062 (4.069) |
| Au | 4.163 | 4.116 | 4.159 | 4.062 (4.065) |
| Pd | 3.949 | 3.913 | 3.943 | 3.876 (3.881) |
| Pt | 3.979 | 3.939 | 3.971 | 3.913 (3.916) |
| Rh | 3.765 | 3.773 | 3.830 | 3.793 (3.798) |
| Ir | 3.873 | 3.844 | 3.871 | 3.831 (3.835) |
4. Adsorption models
Using the respective lattice constants from the PBE, PBE + vdW and PBE + vdWsurf methods, a (3 × 3) unit cell was imposed to represent the periodic (111) surface of each metal. The substrate was modeled by a slab with six atomic layers, and different slabs were separated by 20 Å of vacuum, which ensures that the interaction between the adsorbed molecule and the periodic images of the slab is negligible. The adsorbate and the uppermost two metal layers were allowed to relax, while the bottom four layers were fixed at their bulk positions. We explored the PES of a single Bz by varying the orientation and position of the molecule on top of metal surfaces, followed by geometry relaxation. For the sampling of the Brillouin zone of the (3 × 3) surface, we used a 6 × 6 × 1 Monkhorst–Pack mesh. Figure 1 shows the high-symmetry adsorption sites for Bz on the (111) surfaces of transition metals. We only consider stable adsorption configurations, where the Bz molecule is placed above the metal surfaces with the carbon ring parallel to the substrate [44]. Note that all metals studied here (Cu, Ag, Au, Pd, Pt, Rh and Ir) have a face-centered cubic (fcc) crystal structure, with a three-layer packing sequence of ABCABC. The Bz molecule can thus adsorb at four different sites, labeled as 'atop', 'bri' (abbreviation for 'bridge'), 'fcc' and 'hcp'. Each site has two orientations of 0° and 30°, referring to the angles of the C–C bond relative to the close-packed metal rows [14, 17]. The stability of the adsorption systems is measured by the adsorption energy, Ead, which is defined as Ead = −(EAdSys-EMe-EBz). The subscripts AdSys, Me and Bz denote the adsorption system, the clean metal substrate and the Bz molecule in the gas phase, respectively.
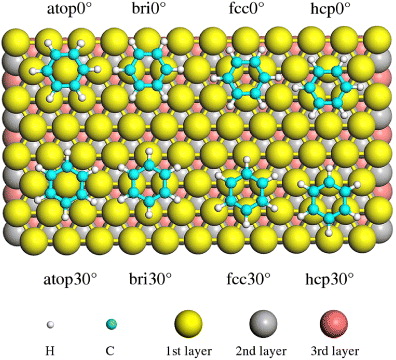
Figure 1. Illustration of the high-symmetry adsorption sites for Bz on the (111) surfaces of transition metals. Due to the fcc structure of Cu, Ag, Au, Pd, Pt, Rh and Ir, there are four possible adsorption sites (atop, bridge, fcc and hcp) for Bz on the (111) metal surfaces. At each site, Bz has two orientations, i.e. 0° and 30°, referring to the angles of the C–C bond being rotated with respect to the neighboring metal–metal bond.
Download figure:
Standard image High-resolution image5. Benchmark calculations for Bz on coinage metal clusters
To assess the accuracy of the DFT + vdWsurf method, we first carry out RPA calculations for Bz on coinage metal clusters, and compare them with the PBE + vdWsurf binding energies. The RPA approach combines the exact exchange energy with an approximate correlation energy at the RPA level. One advantage of the RPA method is that vdW interactions are naturally included in the RPA correlation energy, and that it does not imply any empiricism. Computationally the RPA method is much more demanding than the DFT + vdW method, but has the advantage of more general applicability and also includes the screening from metal electrons automatically. Table 3 shows the binding energy obtained from several approaches, where each system was modeled with a Bz molecule and a cluster of 22 metal atoms applying the PBE + vdWsurf geometry. Testing the basis set dependence of the RPA results showed that the adsorption energy converges for Bz on the Cu22 cluster when using 'tight' settings including the 'tier3' basis set in the FHI-aims code. These settings are thus used consistently for RPA calculations for all systems. The 'EX + cRPA + rSE' scheme corresponds to the treatment of exchange and correlation in terms of 'exact-exchange plus correlation in the random-phase approximation, including the contribution of renormalized single excitations (rSE)'. Our previous work has demonstrated that by adding the rSE contribution to the standard (EX + cRPA)@PBE approach [97], the binding energies in molecules and solids are significantly improved [80]. The standard RPA scheme underestimates the interaction of Bz with the metal surfaces, which was attributed to the too-repulsive nature of the exact-exchange (EX) energies based on PBE orbitals [80]. The DFT + vdWsurf method agrees within 0.1 eV of the (EX + cRPA + rSE)@PBE binding energies for the Ag and Au clusters. PBE predicts a completely repulsive interaction for all the three systems, partly because the metal clusters are constructed based on the PBE + vdWsurf relaxed adsorption structures. Indeed, the PBE adsorption energy increases to 0.01 eV (and close to the slab result) when the Cu cluster is created based on the PBE relaxed periodic slab. Due to the small size of the cluster model used in these calculations, all of the adsorption energies listed in table 3 are significantly smaller than those using periodic systems (table 4). We did not use the most recently developed rPT2 method (renormalized second-order perturbation theory) [98, 99], which includes the RPA, second-order screened exchange (SOSEX) and rSE corrections, to study Bz on coinage metal clusters. This is because SOSEX is a short-range contribution that does not alter the polarizability; thus it has little effect on dispersion interactions [98]. However, SOSEX may well play a role in short-range interactions.
Table 3. Comparison of the calculated adsorption energies Ead (in eV) from different methods for Bz on the Cu22, Ag22 and Au22 clusters. The metal-cluster geometries are constructed by cutting out two-layer-thick segments (14 and 8 atoms in the first and second layer, respectively) from the PBE + vdWsurf relaxed adsorption structures.
| Method | Cu22 | Ag22 | Au22 |
|---|---|---|---|
| (EX+cRPA)@PBE | 0.09 | 0.23 | 0.44 |
| (EX+cRPA+rSE)@PBE | 0.31 | 0.40 | 0.58 |
| PBE+vdWsurf | 0.58 | 0.45 | 0.50 |
| PBE | −0.36 | −0.31 | −0.10 |
Table 4. Calculated adsorption energies Ead (in eV) for Bz/metal at different sites, using the PBE, PBE + vdW and PBE + vdWsurf methods. We placed Bz at each possible adsorption site, and then performed geometry relaxation. A (3 × 3) unit cell is used to represent the periodic (111) surface, with six metallic layers for each slab.
| Site | PBE | PBE+vdW | PBE+vdWsurf | Site | PBE | PBE+vdW | PBE+vdWsurf | ||
|---|---|---|---|---|---|---|---|---|---|
| Cu | atop0° | 0.07 | 0.99 | 0.76 | Ag | atop0° | 0.08 | 0.80 | 0.68 |
| atop30° | 0.07 | 1.00 | 0.84 | atop30° | 0.08 | 0.82 | 0.66 | ||
| bri0° | 0.08 | 1.05 | 0.82 | bri0° | 0.09 | 0.85 | 0.75 | ||
| bri30° | 0.08 | 1.06 | 0.83 | bri30° | 0.09 | 0.85 | 0.75 | ||
| fcc0° | 0.08 | 1.06 | 0.84 | fcc0° | 0.09 | 0.86 | 0.75 | ||
| fcc30° | 0.08 | 1.06 | 0.85 | fcc30° | 0.09 | 0.85 | 0.75 | ||
| hcp0° | 0.08 | 1.07 | 0.84 | hcp0° | 0.09 | 0.87 | 0.75 | ||
| hcp30° | 0.08 | 1.07 | 0.86 | hcp30° | 0.09 | 0.87 | 0.75 | ||
| Au | atop0° | 0.16 | 0.82 | 0.70 | Pd | atop0° | 0.40 | 1.31 | 1.37 |
| atop30° | 0.16 | 0.83 | 0.74 | atop30° a | |||||
| bri0° | 0.15 | 0.84 | 0.73 | bri0° | 0.78 | 1.62 | 1.81 | ||
| bri30° | 0.15 | 0.84 | 0.74 | bri30° | 1.17 | 2.01 | 2.14 | ||
| fcc0° | 0.15 | 0.84 | 0.73 | fcc0° | 0.96 | 1.77 | 1.94 | ||
| fcc30° | 0.14 | 0.83 | 0.74 | fcc30° a | |||||
| hcp0° | 0.15 | 0.85 | 0.74 | hcp0° | 0.99 | 1.81 | 1.97 | ||
| hcp30° | 0.15 | 0.84 | 0.74 | hcp30° a | |||||
| Pt | atop0° | −0.37 | 0.78 | 0.63 | Rh | atop0° b | |||
| atop30° c | atop30° b | ||||||||
| bri0° | 0.17 | 1.14 | 1.28 | bri0° | 0.97 | 2.25 | 2.17 | ||
| bri30° | 0.81 | 1.80 | 1.96 | bri30° | 1.48 | 2.79 | 2.52 | ||
| fcc0° | 0.42 | 1.37 | 1.60 | fcc0° | 1.40 | 2.63 | 2.43 | ||
| fcc30° c | fcc30° b | ||||||||
| hcp0° | 0.50 | 1.45 | 1.68 | hcp0° | 1.46 | 2.71 | 2.51 | ||
| hcp30° c | hcp30° b | ||||||||
| Ir | atop0° | −0.89 | 0.38 | 0.19 | |||||
| atop30° d | |||||||||
| bri0° | 0.36 | 1.42 | 1.43 | ||||||
| bri30° | 1.10 | 2.20 | 2.24 | ||||||
| fcc0° | 0.87 | 1.90 | 1.99 | ||||||
| fcc30° d | |||||||||
| hcp0° | 0.96 | 2.00 | 2.09 | ||||||
| hcp30° d |
aThe atop30°, fcc30° and hcp30° sites convert to the most stable bri30° site after relaxation. bThe atop0° site converts to hcp0°, while atop30°, fcc30° and hcp30° convert to bri30° after relaxation. cThe atop30° and fcc30° sites convert to bri30°, while hcp30° converts to hcp0° after relaxation. dThe atop30°, fcc30° and hcp30° sites convert to bri30° after relaxation.
We conclude that PBE + vdWsurf is a consistent method in comparison to EX + cRPA + rSE, and now we proceed to overview the binding energies, adsorption structures and electronic properties for Bz adsorption on transition-metal surfaces.
6. Estimation of adsorption energies from temperature-programmed desorption spectra
Before proceeding to discuss Bz adsorption systems, in this section we explain our procedure to compute the adsorption energies from TPD measurements, and the range of values for the pre-exponential factors employed in the Redhead formula [40].
The Redhead formula was derived from the kinetic expression for the rate of desorption N(t) [40],

where ν, σ, Hd, kB and Td are the pre-exponential factor, the surface coverage, the desorption enthalpy, the Boltzmann constant and the desorption temperature, respectively. Redhead assumed that Hd is independent of surface coverage and that desorption followed first-order kinetics (n = 1 in equation (1)). Then the above equation can be rearranged as follows [40]:

where β denotes the heating rate. Note that Hd/kBTd values are between 25 and 35 for most TPD spectra taken at near-saturation coverages [100]. Thus the second part in brackets, i.e. ln(Hd/kBTd), is estimated to be 3.64 in the formula [40]. This approximation leads to less than 1.5% error for ν/β between 108 and 1013 K−1 [40].
Henceforth, the pre-exponential factor ν is the only unknown parameter in equation (2), which for very small molecules was determined to be of the order of 1013 s−1. More recent experimental [41–43] and theoretical [101] studies have demonstrated that the ν values for larger molecules are significantly increased compared to the value of 1013 s−1. For example, the prefactors for molecules of the size of Bz are within the range 1015–1018 s−1 [41, 42, 101]. In particular, Campbell and Sellers [43] developed a method to reliably predict the ν values for surface reactions and desorption, by estimating the standard adsorbate entropy, S0ad, and the entropy of gas-phase molecules, S0gas. Using this method, we revisited the adsorption energy values for Bz on coinage metal surfaces, and obtained a prefactor of ∼1015.2 s−1 for Bz [102]. We also note that adsorption enthalpy Had is always larger in magnitude than Hd by kBTd/2 due to the gas impingement rate [32, 103, 104]. On the other hand, the adsorption energy Ead is larger than Had by kBTd since enthalpies (not energies) are measured in experiments [105]. Therefore, one should increase the Hd values by 3kBTd/2 to compare with energy differences calculated by DFT. The reference adsorption energy values reported in table 5 are thus determined from the available TPD measurements using the procedure described above.
Table 5. Comparison of the calculated adsorption energy at the hcp30° site, Ead (in eV), with the results from other theoretical and TPD experiments for Bz on the (111) surfaces of Cu, Ag and Au. The average distances between carbon (or hydrogen) and the first metal layer, dCM (or dHM) (in Å), are also listed and compared with the cited data. lCC denotes the bond length of carbon–carbon in the Bz molecule. lCC1 and lCC2 have the same value when Bz is adsorbed on the coinage metal surfaces.
| Substrate | Method | Ead | dCM | dHM | lCC |
|---|---|---|---|---|---|
| Cu | PBE | 0.08 | 3.74 | 3.74 | 1.40 |
| PBE+vdW | 1.07 | 3.04 | 3.06 | 1.40 | |
| PBE+vdWsurf | 0.86 | 2.79 | 2.79 | 1.40 | |
| GGA | −0.17–0.05 [9, 10, 15, 17] | 3.70 [10] | |||
| MP2 | 0.35 [18] | 3.60 [18] | |||
| vdW-DF | 0.45–0.55 [9, 10, 16, 17] | 3.70 [10, 16, 17] | |||
| M06-L | 0.51 [19] | 3.10 [19] | |||
| DFT-D | 0.61–0.86 [16] | 2.90 [20] | |||
| Experiment | 0.71 [13, 43] | ||||
| Ag | PBE | 0.09 | 3.69 | 3.70 | 1.40 |
| PBE+vdW | 0.87 | 3.14 | 3.15 | 1.40 | |
| PBE+vdWsurf | 0.75 | 2.96 | 2.95 | 1.40 | |
| GGA | −0.16–0.06 [9, 10, 15] | 3.70 [10] | |||
| MP2 | 0.33 [26] | 3.70 [26] | |||
| vdW-DF | 0.41–0.49 [9, 10, 16, 17] | 3.70 [10, 16, 17] | |||
| M06-L | 0.56 [19] | 3.20 [19] | |||
| optB88-vdW | 0.72 [58] | ||||
| DFT-D2 | 1.20–1.29 [28] | 2.75–2.90 [28] | |||
| DFT-D3 | 0.95 [28] | 2.90–3.00 [28] | |||
| Experiment | 0.69 [24, 43] | ||||
| Au | PBE | 0.15 | 3.62 | 3.62 | 1.40 |
| PBE+vdW | 0.84 | 3.21 | 3.20 | 1.40 | |
| PBE+vdWsurf | 0.74 | 3.05 | 3.04 | 1.40 | |
| GGA | −0.15–0.08 [9, 10, 15] | 3.70 [10] | |||
| MP2 | 0.31 [26] | 3.80 [26] | |||
| vdW-DF | 0.42–0.55 [9, 10, 16, 17, 30] | ||||
| M06-L | 0.55 [19] | 3.20 [19] | |||
| optB88-vdW | 0.79 [58] | 3.23 [58] | 3.23 [58] | ||
| DFT-D | 0.76–1.35 [16] | 3.10 [20] | |||
| Experiment | 0.76 [29, 43] | ||||
7. Overview of Bz adsorption on metals
The different bonding nature for weakly bound systems (Bz on Cu, Ag and Au) and strongly bound systems (Bz on Pd, Pt, Rh and Ir) is clearly reflected in the adsorption energies listed in table 4. The interaction between Bz and the coinage metal surfaces is much weaker than for Bz and the transition metals with only partially filled d bands. For example, our PBE + vdWsurf calculations predict a flat PES for all physisorption systems, and this observation agrees with scanning tunneling microscopy (STM) experiments which showed that Bz molecules can diffuse freely over Cu(111) and Au(111) terraces at low temperatures (4 or 77 K) [106–108]. On the reactive Pt(111) surface, Bz was found to chemisorb at the bridge site, with an angle of 30° between the C–C and Pt–Pt bonds, based on the low-energy electron diffraction (LEED) analysis [36]. Our PBE + vdWsurf calculations indicate that Bz adsorbs at the bri30° site not only for Bz/Pt(111), but also for other covalently bound systems Pd(111), Rh(111) and Ir(111), with a relatively larger energy corrugation than in the case of coinage metals (e.g. 1.33 eV for Bz on the Pt(111) surface). We also note that the average distances between carbon and Pd, Pt, Rh and Ir are almost identical (in the range of 2.08–2.13 Å from PBE + vdWsurf; table 6). The adsorption sites hcp30° for Bz on Cu(111), Ag(111) and Au(111); bri30° for Bz on Pd(111), Pt(111), Rh(111) and Ir(111) chosen to show the results in figures 2 and 3 correspond to the minima for the calculated PES. As shown in figure 2, PBE yields negligibly small adsorption energies for Bz on coinage metal surfaces, while PBE does bind, but not sufficiently strongly for Bz on the Pd, Pt, Rh and Ir surfaces. Upon including the vdW energy, the PBE + vdW method significantly enhances the binding energies for all the studied systems. In addition, the collective response of the substrate electrons reduces the vdW coefficients and vdW radii for all metals (table 1), leading to opposing effects in the vdW energy and resulting in non-trivial behavior for different metals.

Figure 2. Adsorption energies Ead (in eV) and the average distance between carbon and the first metal layer dCM (in Å) for Bz on the (111) surfaces of Cu, Ag, Au, Pd, Pt, Rh and Ir from the PBE, PBE + vdW and PBE + vdWsurf methods.
Download figure:
Standard image High-resolution image
Figure 3. (a) Side and top views of the adsorption structure at the hcp30° site for Bz on the (111) surfaces of Cu, Ag and Au. (b) Adsorption structure at the bri30° site for Bz on the (111) surfaces of Pd, Pt, Rh and Ir. dCM (dHM) indicates the average distance between carbon (hydrogen) atoms and the first metal layer. lCM and lCC denote the bond lengths of carbon–metal and carbon–carbon, respectively. The adsorbed Bz is flat-lying above the coinage metal surfaces, whereas it deforms significantly upon adsorption on the Pd, Pt, Rh and Ir surfaces.
Download figure:
Standard image High-resolution imageTable 6. Comparison of the calculated adsorption energies at the bri30° site, Ead (in eV), with the results from other theoretical calculations and experiments for Bz on the (111) surfaces of Pd, Pt, Rh and Ir. The average distances between carbon (or hydrogen) and metals in the first layer, dCM (or dHM) (in Å), are also listed in the table. The average bond lengths between carbon and the nearest metal atoms, lCM (in Å), are measured in the equilibrium structures and compared with data from the literature. lCC denotes the carbon–carbon bond length in the Bz molecule.
| Substrate | Method | Ead | dCM | dHM | lCM | lCC1 | lCC2 |
|---|---|---|---|---|---|---|---|
| Pd | PBE | 1.17 | 2.12 | 2.47 | 2.21 | 1.45 | 1.43 |
| PBE+vdW | 2.01 | 2.12 | 2.47 | 2.22 | 1.45 | 1.43 | |
| PBE+vdWsurf | 2.14 | 2.10 | 2.46 | 2.20 | 1.45 | 1.43 | |
| optB88-vdW | 1.91 [58] | ||||||
| GGA | 1.04–1.19 [60, 61, 63] | 2.23 [63] | 1.45 [63] | 1.43 [63] | |||
| Pt | PBE | 0.81 | 2.10 | 2.54 | 2.18 | 1.47 | 1.43 |
| PBE+vdW | 1.80 | 2.11 | 2.53 | 2.19 | 1.48 | 1.44 | |
| PBE+vdWsurf | 1.96 | 2.08 | 2.51 | 2.18 | 1.48 | 1.43 | |
| optB88-vdW | 1.84 [58] | 2.12 [58] | 2.53 [58] | ||||
| GGA | 0.85–1.21 [60, 61, 63–66] | 2.17 [62] | 2.22 [61, 63] | 1.47 [63] | 1.43 [63] | ||
| Experiment | 1.57–1.91 [32] | 2.02±0.02 [36] | |||||
| Rh | PBE | 1.48 | 2.14 | 2.55 | 2.18 | 1.47 | 1.44 |
| PBE+vdW | 2.79 | 2.14 | 2.53 | 2.18 | 1.47 | 1.44 | |
| PBE+vdWsurf | 2.52 | 2.12 | 2.51 | 2.20 | 1.47 | 1.43 | |
| optB88-vdW | 2.27 [58] | ||||||
| GGA | 1.53 [63] | 2.20 [63] | 1.46 [63] | 1.43 [63] | |||
| Ir | PBE | 1.10 | 2.15 | 2.60 | 2.20 | 1.48 | 1.44 |
| PBE+vdW | 2.20 | 2.14 | 2.59 | 2.20 | 1.48 | 1.44 | |
| PBE+vdWsurf | 2.24 | 2.13 | 2.57 | 2.19 | 1.48 | 1.44 | |
| optB88-vdW | 2.09 [58] | ||||||
The computed adsorption energies exhibit several interesting trends. Despite the fact that the C6 coefficients and vdW radii are essentially the same for Pt and Au [76], the adsorption-energy difference between PBE + vdWsurf and PBE is significantly larger for Bz/Pt(111), a typical covalently bound system, than for Bz/Au(111), a typical physisorbed system (1.15 versus 0.59 eV). This is a remarkable finding because vdW interactions are typically expected to play a minor role in strongly bound, chemisorbed systems [58].
We now discuss the reason for the larger vdW energy contribution in Bz/Pt(111) when compared to Bz/Au(111). Figure 4 shows the vdW energy contribution to the adsorption energy distributed as a function of the distance between the molecule and the metal. Here in the top panel of figure 4 the vdW energy at a distance R refers to the sum of all interatomic vdW energies between the atoms in Bz and the metal atoms that are R Å apart from each other. The vdW energy histogram in the top panel demonstrates that a significant part of the vdW energy arises from a rather short distance range of 3–4 Å for both metals. As the Bz molecule adsorbs much closer to the Pt surface than to the Au surface, the vdW interaction is greater for Bz/Pt(111). This fact is further demonstrated by the onset of the vdW energy for Pt, which starts 0.5 Å closer than for the Au surface. The integrated vdW interaction (bottom panel) exemplifies the faster increase of the vdW energy for Bz/Pt(111) as a function of the Bz–metal distance. For both cases, the vdW energy essentially converges within 1.5 meV of the periodic limit beyond ∼15 Å. The difference in vdW energy between Pt and Au converges at around 8 Å, as represented in the inset, as the long-range vdW coefficients are essentially the same for Bz/Pt(111) and Bz/Au(111). The greater role of vdW energy in strongly bound systems can also be generalized to other transition-metal surfaces [58].
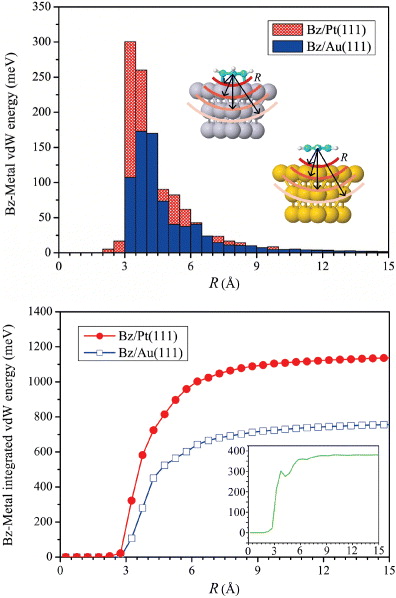
Figure 4. Top: the vdW energy for Bz on Pt(111) and Au(111) as a function of radial distance, R, from the PBE + vdWsurf method. Bottom: the integrated vdW energy of Bz–metal versus R. The inset plot shows the difference between the vdW energy on Pt(111) and Au(111).
Download figure:
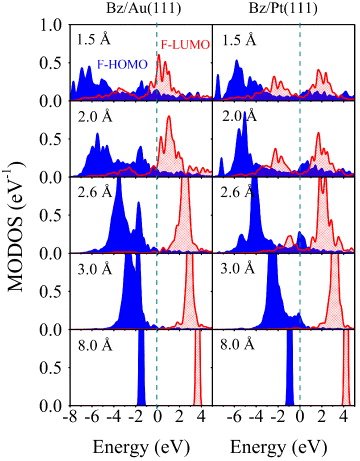
Standard image High-resolution imageThe different nature of bonding in the physisorption and covalently bound cases is also distinguished in the molecular orbital density of states (MODOS). The MODOS was calculated by projecting the total density of states of the full system onto the HOMO and LUMO orbitals of the free molecule using the same geometry of the molecule adsorbed on the surface [109]. As shown in figure 5, the molecular bands broaden as the molecule approaches the substrate, and at 3.0 Å, around the equilibrium distance of Bz/Au(111), the broadening of the former HOMO and LUMO (F-HOMO and F-LUMO) is discernible for Bz on Au and on Pt. Almost zero charge transfer is obtained for Bz/Au(111), which is consistent with the physisorptive nature of bonding between Bz and the Au(111) surface. For Bz/Pt(111) at 3.0 Å a small portion of the F-HOMO density of states (DOS) lies above the Fermi level, which reflects small electron transfer from the adsorbate to the metal. The electron transfer is further increased for Bz/Pt(111) at a distance of 2.6 Å. A prominent feature in this case is that the F-LUMO orbital is shifted below the Fermi level, indicating back-donation from the metal to the adsorbate. At a distance of 2.6 Å, the broadening of the HOMO and LUMO is already visible. When the Bz molecule gets closer to the substrate (2.0 Å) this broadening becomes significant, and at the 'extremely' close distance of 1.5 Å, the energy range of the two MODOSs is practically the same.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. MODOS projected on the free Bz molecule HOMO and LUMO orbitals (F-HOMO and F-LUMO) as a function of adsorption height for the Bz/Au(111) (left panel) and Bz/Pt(111) systems (right panel). Each structure was relaxed by the PBE + vdWsurf method, with the z-coordinate of the carbon backbone fixed. The zero of energy corresponds to the Fermi level.
Download figure:
Standard image High-resolution image{kind=link}
Based on the calculated adsorption energies, the binding structures and the electronic properties, the Bz/metal systems can be divided into two categories: (i) weak adsorption, including Bz on Cu(111), Ag(111) and Au(111); and (ii) strong adsorption, including Bz on Pd(111), Pt(111), Rh(111) and Ir(111). In the following sections, we will provide more detailed analysis for benzene adsorbed on transition-metal surfaces, in order to further clarify the critical role of vdW interactions in the prediction of adsorption structures and energetics, and also to deepen our understanding of the nature of bonding for the adsorption of aromatic molecules. For each system, we start by briefly reviewing the previous experimental and theoretical work. Then we analyze the PES by comparing adsorption energies at eight high-symmetry adsorption sites. For the most stable site, we further discuss the impact of pairwise and collective vdW interactions on adsorption heights and binding energies. We also compare PBE + vdWsurf results with the available experiments and other theoretical work.
8. Weak adsorption: Bz at Cu(111), Ag(111) and Au(111)
8.1. Previous studies
Adsorption of Bz at the Cu(111), Ag(111) and Au(111) surfaces is weak because their d-bands are well below the Fermi level and full. Thus, metal d-states participate only weakly in the bonding [33]. This also implies that the energy corrugation for Bz adsorption is very shallow. In earlier experiments, Xi et al [13] investigated the structure of Bz on the Cu(111) surface using high-resolution electron energy loss spectroscopy (HREELS) and near-edge x-ray absorption fine structure (NEXAFS) measurements. They found that Bz in the first layer binds parallel to the Cu(111) surface. Their TPD experiments revealed that at a heating rate of 4 K s−1, Bz desorbs from Cu(111) at a low temperature of 225 K, with a high-temperature tail extending to 300 K [13]. Lukas et al [12] reproduced the TPD experiments and reported that Bz has a stronger binding at step edges and point defects than on the clean surface. Yannoulis et al [110] studied the orientation of ordered Bz/Ag(111) layers using NEXAFS. They reported a flat bonding geometry. By means of angle-resolved photoemission spectroscopy in combination with LEED, Dudde et al [111] studied a (3 × 3) overlayer of Bz adsorbed on Ag(111). They concluded that the Bz molecules occupy the three-fold hollow sites of the Ag(111) surface, but it remained unclear whether the molecules occupy fcc or hcp hollow sites. Such a three-fold hollow site has also been reported by Moskovits and DiLella [112] using surface-enhanced Raman spectroscopy, and by Avouris and Demuth using the HREELS method [113]. Zhou et al [24] found that Bz desorbs from Ag(111) at 220 K, i.e. at a slightly lower temperature than for the Bz/Cu(111) system [12, 13]. Syomin et al [29] showed that Bz at Au(111) desorbs at 239 K at a low coverage of 0.1 ML. They also found that the desorption temperature decreases with increasing coverage, and a broad Bz desorption feature occurs at 210 K after completion of the monolayer.
The standard DFT-GGA calculations for the adsorption of aromatic molecules on metals have been systematically reviewed by Jenkins [33]. Due to the importance of vdW forces in physisorptive systems, several 'beyond-GGA' approaches have been employed to study the interactions of Bz on coinage metal surfaces. Caputo et al [26] studied Bz/Cu(111) at the level of the second-order perturbation theory (MP2), and illustrated a vdW-type potential energy curve with a shallow minimum. By means of the vdW-DF functional, Berland et al [17] demonstrated that inclusion of vdW interactions enhances the binding energy by an order of magnitude for Bz on Cu(111). They also observed that Bz molecules move almost freely over the fcc, bridge and hollow sites. Using the same method, Toyoda et al [10] and Kelkkanen et al [9] showed that accounting for vdW forces dramatically strengthens the interaction of Bz with the substrates, whereas the equilibrium structures remain unchanged. Tonigold and Gross [16] carried out DFT-D calculations for the weakly bound systems, where the C6 coefficients for metals were either adopted from Grimme's scheme [57, 114] or calculated using a hybrid QM:QM procedure. In the latter approach, the C6 coefficient is determined via a least-squares fitting of the dispersion expression to the adsorption energy difference by MP2 and PBE. The meta-GGA (M06-L) functional was employed by Ferrighi et al [19] to investigate the structure, energetic and electronic properties for Bz adsorption on noble metal surfaces. The general finding of all these 'beyond-GGA' approaches is that they significantly reduce the molecule–surface distance and increase the binding energy.
8.2. Bz on Cu(111)
Using the (3 × 3) supercell, we start by exploring the PES of a single Bz molecule on the metal surface. The calculated adsorption energies for eight high-symmetry sites are listed in table 4. For Bz/Cu(111), vdW forces enhance the binding energy by about 1 eV compared with PBE. Due to the dramatic reduction of the dispersion coefficients used in the PBE + vdW and PBE + vdWsurf methods (253 versus 59 hartree bohr6; table 1), the collective effects from the bulk Cu electrons reduce the PBE + vdW adsorption energy by more than 0.2 eV. Note that the vdW radius R0 of Cu also reduces from 3.76 to 2.40 bohr after accounting for the collective response of the substrate electrons, resulting in compensation for the computed adsorption energy in the Bz/Cu(111) system. Our PBE + vdWsurf results demonstrate that the atop0° site is the least stable adsorption site, while the hcp30° site is slightly more stable than any of the other structures. Despite the profound difference in the magnitude of the computed adsorption energies, the PBE, PBE + vdW and PBE + vdWsurf methods all predict a small energy corrugation for Bz on Cu(111), further supporting the STM observations that Bz can diffuse freely over the Cu(111) surface at 77 K [108]. The flat PES and the mobility of the Bz molecule make it impossible to unambiguously identify the most stable adsorption site. In this work, we select the hcp30° site to perform further structure and energetic analysis for Bz on Cu(111), and we also do so for Ag(111) and Au(111) in the following two subsections. We chose this site mainly because (a) experiments have reported that the three-fold hollow site is the stable site for Bz on coinage metal surfaces [111–113]; and (b) the hollow sites (both fcc and hcp) have already been used as a model system in previous theoretical studies [10, 107].
As exemplified in the Bz/Cu(111) system, the 'absolutely' flat molecular plane is clearly illustrated by the small difference (less than 0.02 Å) between the carbon–metal and hydrogen–metal adsorption heights (see table 5). Including vdW interactions reduces the average C–Cu adsorption height from 3.74 to 3.04 Å. This height further decreases to 2.79 Å after accounting for the collective response of the metal electrons. Due to the similar vdW radius of Cu, the distance predicted by the PBE + vdWsurf method is close to that of DFT-D (2.79 versus 2.90 Å) [20]. The computed workfunction at the above distance agrees well with experiments [20], which in turn validates our computed adsorption height for Bz/Cu(111). We note that the adsorption energies calculated with the vdW-DF functional are the same as the PBE results for all coinage metal surfaces [10, 16, 17]. This was attributed to the overly repulsive exchange part used in the vdW-DF functional, as recently discussed in [94, 115]. The MP2 method gives an adsorption height of 3.60 Å, and the overestimation probably stems from the small Cu cluster used in the calculations [18].
For Bz/Cu(111) at the hcp30° site, the PBE + vdW adsorption energy is almost 1 eV larger than the corresponding PBE result, and the PBE + vdWsurf result (0.86 eV) agrees better with experiments (0.71 eV) [12, 13]. MP2 is known to overestimate the dispersion interactions [116] and yields considerable errors for vdW-bonded systems [73]. However, probably due to the small cluster utilized in the simulation, the MP2 adsorption energy is half of the TPD results [18]. The vdW-DF method provides a lower energy range [9, 10, 16, 17] than the experiments, and this energy difference may derive from the chosen GGA exchange functional [9, 10]. The DFT-D adsorption energies are reported to be 0.61 and 0.86 eV, depending on whether the C6 coefficients are deduced using MP2 or free-atom properties, respectively [16].
8.3. Bz on Ag(111)
The relaxed Bz molecule adsorbs in a flat-lying geometry on the Ag(111) surface, which is consistent with the observations and conclusion from NEXAFS, EELS and Raman spectroscopy [110, 111]. The PBE + vdW method, which enhances the adsorption energy by at least 0.7 eV with respect to PBE, demonstrates that the bridge and hollow sites are more stable than the atop site. The conclusion still holds with the PBE + vdWsurf calculations, although including the collective response of the substrate electrons leads to a consistent decrease in binding. A shallow PES is predicted upon Bz adsorption on Ag(111) and, at all adsorption sites, the Bz–Ag interactions are weaker than those of Bz–Cu (cf table 4).
Analysis of the relaxed structure at the hcp30° geometry shows that there is no adsorption-induced deformation of the Bz molecule. The carbon atoms are only 0.01 Å above or below the hydrogens, and the C–C bond lengths remain unchanged (table 5). The PBE + vdW method decreases the Bz adsorption height by 0.55 Å relative to the PBE result. Collective vdW effects captured by the PBE + vdWsurf method further decrease the distance due to the significant reduction of the vdW radius for Ag (table 1). The carbon–metal distance is larger for Ag than for Cu, which suggests a weaker interaction for the former. The distances from the M06-L functional [19] are close to the PBE + vdW results, but are 0.50 Å shorter than those from PBE. The vdW-DF and MP2 approaches give the same adsorption height as PBE [10, 26].
The adsorption energies for Bz on Cu(111) and Bz on Ag(111), despite being similar to PBE, differ significantly (0.1–0.2 eV) when the PBE + vdW and PBE + vdWsurf methods are used (table 5). In both vdW-inclusive methods, interactions of Bz–Ag are weaker than those of Bz–Cu. This is in agreement with TPD experiments which showed that the Bz molecule desorbs at a lower temperature from Ag(111) than from Cu(111) [12, 13, 24]. PBE + vdWsurf decreases the Bz/Ag(111) adsorption energy (0.75 eV) and yields much better agreement with TPD experiments (0.69 eV) [24]. The adsorption energy from the optB88-vdW approach is also close to the experiments [58, 69]. In contrast, the binding of Bz to the Ag(111) surface is substantially underestimated by the MP2, M06-L and vdW-DF methods [9, 10, 19, 26].
8.4. Bz on Au(111)
The flatness of the PES for Bz on Au(111) which results from our calculations with the PBE, PBE + vdW and PBE + vdWsurf methods confirms the STM observations that Bz molecules are mobile over Au(111) terraces even at 4 K [107]. PBE + vdW predicts that the bridge and hollow sites have similar adsorption energies in the range of 0.82–0.85 eV (table 4). PBE + vdWsurf lowers the energy by about 0.1 eV with respect to the PBE + vdW result. An almost identical adsorption energy is found for all sites, which indicates a small barrier for surface diffusion of Bz on the Au(111) surface. The Bz/Au(111) adsorption energies are lower than those of Bz/Cu(111), but are close to the Bz/Ag(111) results.
As summarized in table 5, the computed adsorption heights for Bz/Au(111) have the sequence of PBE > PBE + vdW > PBE + vdWsurf at the hcp30° site. The collective response of the metal electrons leads to smaller structural variation for Bz/Au(111) than for Bz/Cu(111), but still changes the equilibrium distance from 3.21 to 3.05 Å. The Bz–Au separation distance was deduced to be 2.95–3.10 Å, based on the experimental workfunction of Bz/Au(111) as well as the adsorption height of pentacene/Au(111) [10, 106, 117]. The PBE + vdWsurf method shows the best performance compared to the deduced experimental data. Larger than 5%, but less than 10% errors are found for M06-L [19] and DFT-D [20], while an error of more than 25% is obtained when the PBE (3.70 Å) [10], vdW-DF (3.70 Å) [10] and MP2 (3.80 Å) [26] methods are used.
The PBE + vdWsurf adsorption energy for Bz/Au(111) (0.74 eV) is in excellent agreement with the TPD experiments (0.76 eV) [29] and the predictions from the optB88-vdW functional (0.79 eV) [58] and the hybrid QM:QM DFT-D method (0.76 eV) [16]. Due to the overestimation of the C6 coefficients for heavier atoms, the DFT-D adsorption energy is significantly larger when the vdW parameters were obtained by Grimme's method (1.35 eV) [16]. The vdW-DF adsorption energies (0.42–0.55 eV) [9, 10, 30] are too small compared with both the PBE + vdWsurf data and the TPD values with updated pre-exponential factors. Similarly, the MP2 and M06-L methods underestimate the experimental adsorption energy (see table 5). We also find that the calculated adsorption energy varies with the coverage of the Bz molecule: when using a larger Au(111) surface with a (4 × 4) unit cell, the adsorption energy decreases by 0.12 eV when using the PBE + vdWsurf method.
9. Strong adsorption: Bz at Pd(111), Pt(111), Rh(111) and Ir(111)
9.1. Previous studies
Palladium is among the most important materials in heterogeneous catalysis. However, few experiments have been carried out to study the adsorption of Bz on the Pd(111) surface. Using x-ray photoelectron spectroscopy and NEXAFS, Lee et al [118] reported that the Bz molecule lies flat and binds strongly with the Pd(111) surface at low coverages (below 0.16 ML, molecules per Pd atom). They also found that Bz tilts with respect to the substrate at coverages above 0.16 ML. Waddill and Kesmodel [119] concluded from their HREELS and TPD experiments that the Bz rings locate at only one site and parallel to the Pd(111) surface. The interaction of Bz with the Pt(111) surface, including adsorption and dehydrogenation, has been extensively studied among the Bz adsorption systems. Nevertheless, even the preferred adsorption site remains controversial in experiments. By means of the diffuse LEED intensity analysis, Wander et al [36] reported that the bri30° site is the most stable site for Bz chemisorbed on the Pt(111) surface. In contrast, Tirendi et al [120] used nuclear magnetic resonance and suggested that Bz molecules are located at the atop site. Inferred from the orientations of the STM images, Weiss and Eigler [121] reported the coexistence of Bz molecules at both the hcp and fcc sites. Despite the ambiguous adsorption site, all experiments clearly conclude that the adsorbate lies flat on the Pd(111) surface, binding with the Bz π orbitals to the Pt d bands. The STM images suggest immobile Bz molecules adsorbed on Pt(111), which reflects the strong binding [121, 122]. The Bz molecules are found to adsorb as intact molecules on the Pt(111) surface at 300 K [32]. However, for coverages below 0.6 ML, Bz dissociates completely into hydrogen gas and adsorbed graphitic carbon upon heating [32]. Therefore, microcalorimetric measurements, rather than desorption-based methods (such as TPD, molecular beam relaxation spectroscopy and equilibrium adsorption isotherms), are required for determining the heat of adsorption for Bz on the Pt(111) surface. Sautet and Joachim [123] simulated the STM images for Bz on Rh(111), and achieved good agreement with the STM experiments carried out by Ohtani et al [124]. Molecular distortion of Bz, including both planar and buckling distortions, has been observed on Rh(111) by means of a LEED intensity analysis [125]. Scarce previous work has focused on the Bz/Ir(111) system. Using angle-resolved photoelectron spectroscopy and electron-stimulated desorption ion angular distribution, Mack et al [126] reported the trigonal distortion of Bz molecules in a planar adsorption geometry on the Ir(111) surface.
From the modeling point of view, DFT-GGA calculations using PBE and PW91 functionals have been carried out to study the coverage-dependent adsorption energies, molecular distortions and STM images for Bz on the Pd(111), Pt(111) and Rh(111) surfaces [60, 63, 127–129]. The predicted adsorption sites for these chemisorption systems are consistent with the experimental HREELS conclusion that Bz molecules adsorb predominantly at the bridge site of Pt(111), while they can adsorb at the bridge and hollow sites of Pd(111) and Rh(111) [63].
9.2. Bz on Pd(111)
The calculated adsorption energies show, not surprisingly, that the interaction of Bz with Pd(111) is much stronger than for coinage metal surfaces (see table 4). The bri30° and hcp0° sites are the two most stable sites among the eight studied possibilities. The fcc0° site is slightly less stable than the hcp0° site, but it has a more than 0.5 eV higher adsorption energy than the atop0° site. At each site the PBE + vdW method increases the PBE adsorption energies by more than 0.80 eV, while PBE + vdWsurf further increases the adsorption energies by 0.15 eV.
The bri30° site, which has the strongest binding, is used as the model system to carry out further structural analysis. The nature of covalent bonding is explicitly demonstrated in figure 3(b) by the small separation distance and large deformation of the adsorbed Bz molecule. The carbon ring is parallel to the substrate and the hydrogen atoms tilt upward after relaxation. The vertical distances between the carbon and hydrogen atoms are 0.35–0.36 Å from PBE, PBE + vdW and PBE + vdWsurf (table 6). The C–C bonds elongate upon Bz adsorption on the Pd(111) surface, where the carbon bonds across the Pd–Pd bond (lCC1) are slightly longer than those above the Pd atom (lCC2). The deformation in the adsorbed Bz and the substrate leads to a large distortion energy of 0.89 eV [63].
The adsorption energies for Bz/Pd(111) at the bri30° site reveal that vdW forces play an unexpectedly strong role in Bz chemisorption (see table 6). The binding energy increases significantly from 1.17 eV (PBE) to 2.01 eV (PBE + vdW). We have also tested the role of collective response of the substrate electrons inside the Pd(111) bulk, finding adsorption energy differences of 0.13 eV, with respect to the PBE + vdW result. The PBE + vdWsurf method enhances the binding energy compared with PBE + vdW, which is rationalized by the fact that the screening inside the Pd(111) substrate is much smaller than for coinage metals (35% reduction in the Pd–Pd C6 coefficient and 16% reduction in the vdW radius). Similar results have also been found in our recent work on isophorone molecule adsorption on the Pd(111) surface [130]. The screening effect is smaller for Pd than for Ag, which is presumably because Ag has a partially filled 5s orbital, while the 5s orbital of Pd is (nearly) empty. Note that for Pd bulk the d-band is essentially full. Thus, d-orbitals are expected to play a smaller role than in other transition metals with partially occupied d-bands.
9.3. Bz on Pt(111)
For Bz on the Pt(111) surface, the most favorable adsorption energy is predicted at the bri30° site for all studied functionals: PBE (0.81 eV), PBE + vdW (1.80 eV) and PBE + vdWsurf (1.96 eV) (cf table 4). The second and third preferred sites are the hcp0° and fcc0° sites, respectively. The distortion energy due to geometry change in the Bz molecule and the substrate surface in the Bz/Pt(111) system is a factor of two larger than that in Bz/Pd(111) (1.84 versus 0.90 eV) [63]. Also the vertical distance between carbon and hydrogen for Bz on Pt(111) is almost 0.1 Å greater than that for Bz on Pd(111) (table 6). Our calculated adsorption height from PBE + vdWsurf is in excellent agreement with those of a LEED analysis (2.08 versus 2.02 ± 0.02 Å) [36].
The Bz/Pt(111) system has been extensively studied by theory [58, 60, 61, 63–66], but most of the previous work neglected the vdW interactions for this system. As shown in table 6, the adsorption energies calculated by the PBE + vdWsurf and optB88-vdW methods match the microcalorimetry result at 0.7 ML (1.57–1.91 eV [32], at the same coverage used for our DFT calculation). We also constructed a larger supercell of (4 × 4) for Pt(111), and in this case the adsorption energy is determined to be 2.18 eV from PBE + vdWsurf, also within the uncertainty of calorimetry measurements in the limit of zero coverage (1.84–2.25 eV) [32]. Further enlarging the size of the unit cell to (5 × 5) leads to only a 0.02 eV difference in the adsorption energy, with respect to the (4 × 4) unit cell.
9.4. Bz on Rh(111)
In contrast to Bz/Pd(111) and Bz/Pt(111), the adsorption energies for Bz adsorbed on the bri30°, hcp0° and fcc0° sites are close for Bz/Rh(111) (see table 4). For example, PBE + vdWsurf predicts that the binding at the bridge site is only 0.01 eV stronger than at the hcp0° site. Since the three geometries have essentially the same energy, one may expect that the Bz molecule can diffuse on the Rh(111) surface. We note that the C6 value has the biggest reduction for Rh (a factor of 5.5; table 1), which can be explained because Rh possesses the largest amount of metallic electrons among the seven transition metals [131, 132]. Consequently, Bz on Rh(111) has the most visible change in the adsorption energy when the PBE + vdWsurf method is employed. The calculated adsorption energies are listed in table 6. They show that the interaction of Bz with Rh(111) is 0.28 eV stronger than with the Pd(111), Pt(111) and Ir(111) surfaces. A similar conclusion has also been obtained when using the optB88-vdW functional [58].
We continue to use the bri30° site to perform the structural analysis. As shown in table 6, the average C–Rh distance is 0.39 Å shorter than the H–Rh distance, which indicates large deformation upon Bz adsorption. In fact, the distortion energy for Bz/Rh was determined to be 1.55 eV, due to large deformation from both the adsorbate and substrate [63]. Including vdW interactions and collective screening effects on top of our PBE calculations leads to only 0.02 Å variation in the adsorption height, although the adsorption energy changes dramatically.
9.5. Bz on Ir(111)
Like all other chemisorbed systems discussed in this work, the bri30° site is the most stable adsorption site for Bz/Ir(111). The hcp0° and fcc0° sites are 0.1–0.2 eV less stable than the bri30° site. PBE gives repulsive interaction for Bz at the atop0° site: the Ir atom connecting with the carbons moves inward by 0.34 Å, relative to the neighboring metal atoms in the same layer. Including the vdW contributions increases the adsorption energy to 0.38 eV at the atop0° site. However, the adsorption energy at atop0° is still at least 1 eV weaker than other high-symmetry adsorption sites. The collective response of the metal electrons does not modify the adsorption energy for the Bz/Ir(111) system.
The relaxed structure shows strong deformation for Bz upon adsorption on Ir(111). The hydrogen atoms tilt and the vertical difference between carbon and hydrogen atoms is 0.44–0.45 Å from the PBE, PBE + vdW and PBE + vdWsurf methods (see table 6). The average distance between carbons and the first metal layer is 2.14 Å, which is more than 1.2 Å shorter than the average height between graphene and the Ir(111) surface (3.41 Å from vdW-DF and 3.38 ± 0.04 Å from experiments) [133]. Due to the large separation distance in the graphene/Ir(111) system, the adsorption energy of Bz/Ir(111) (2.24 eV from PBE + vdWsurf) is much stronger than the graphene/Ir(111) system with 0.05 eV per carbon atom [133].
10. Conclusions
In summary, DFT calculations including the long-range contributions to the vdW interactions and collective response of the substrate electrons within the metal bulk were carried out to investigate the structure and energetic properties of benzene adsorbed on transition-metal surfaces. According to our calculations, the following conclusions were drawn:
- 1.Bz molecules adsorb in a flat arrangement on all the metal surfaces, with weak (physisorption) interaction (0.74–0.86 eV) and almost zero distortion for the noble metals Cu(111), Ag(111) and Au(111), and stronger interaction (1.96–2.52 eV) and large deformation for the 3d and 4d transition metals Pd(111), Pt(111), Rh(111) and Ir(111).
- 2.The PES for Bz on noble metals shows negligible corrugation. In contrast, the bri30° site is clearly the preferred adsorption site for Bz on the (111) surfaces of Pd, Pt and Ir. The energy difference between bri30° and hcp0° is only 0.01 eV for the Bz/Rh(111) system, which suggests the coexistence of the bridge and hollow sites, and sliding of Bz on the Rh(111) surface.
- 3.The vdW interactions play a crucial role not only in the weakly bound systems, but also in the strongly bound systems. For Bz on Cu(111), Ag(111) and Au(111), the adsorption energies from PBE + vdW are at least 0.7 eV larger than those calculated by PBE, while the adsorption heights are decreased by at least 0.4 Å. For strongly bound systems, the PBE + vdW method enhances the adsorption energy by a factor of two, but changes the binding structures only slightly.
- 4.The collective response effects captured via the vdWsurf method significantly reduce the C6 coefficients (by a factor of 1.5–5.5) and vdW radii for bulk coinage metals compared to using free-atom reference vdW parameters. This leads to important reductions both in the adsorption energy and in the equilibrium distance for Bz on Cu(111), Ag(111) and Au(111), with respect to PBE + vdW results. The collective screening effects also play a significant role in the strongly bound systems, leading to a modification of the adsorption energies for Bz on the Pd(111), Pt(111) and Rh(111) surfaces. The Bz/Rh(111) adsorption energy computed by PBE + vdWsurf is 0.27 eV weaker than that of PBE + vdW.
Overall, we demonstrate that the accurate treatment of collective response effects by the substrate electrons is crucial for reliable determination of adsorption structures and energetics of molecules adsorbed on transition-metal surfaces. The remarkable qualitative and quantitative accuracy obtained from PBE + vdWsurf calculations in modeling the benzene/metal interfaces suggests that this method can be used for studying more complex organic molecules on metal surfaces, as already demonstrated in recent work [58, 76–79]. We emphasize that the DFT + vdWsurf method does not depend on the nature of the substrate surface, being equally applicable to insulators, semiconductors and metals. Ongoing applications of the DFT + vdWsurf method include adsorption on non-close-packed surfaces, defects and other complex adsorption scenarios.
Acknowledgments
We are grateful for support from the FP7 Marie Curie Actions of the European Commission, via the Initial Training Network SMALL (MCITN-238804). WL was funded by a fellowship from the Alexander von Humboldt Foundation. AT acknowledges support from the European Research Council (ERC Starting Grant VDW-CMAT). BS is supported by Scientific Discovery through Advanced Computing (SciDAC) program DE-SC0008626.