
Abstract
Carbyne and carbyne-based low-dimensional structures are promising for several applications including ultra-compact circuits and purification devices. Designing any applied carbyne-based structure requires a fundamental understanding of the mechanical strength of carbyne chains with different lengths at different temperatures and operating chemical environment. Here we use molecular dynamics simulations to investigate the strength of carbyne chains with different lengths at different temperatures. A theoretical framework based on statistical mechanics and molecular dynamics results is presented, proving a fast and insightful method for predicting the rupture force and its physical mechanism. The effect of water molecules' interaction is also studied on the mechanical properties and it is shown that both the tensile strength and rupture strain are improved by the water interaction. The results of this work can be used for designing and analyzing the robustness and reliability of various carbyne-based materials and applied devices for varies working conditions.
Export citation and abstract BibTeX RIS
1. Introduction
Carbyne is a one-dimensional carbon allotrope composed of sp-hybridized carbon atoms, which in theory consists of either double-bonded structure (polycumulene) or alternating single-triple bonds (polyyne) [1–3]. Several chemical methods were proposed for synthesizing stable finite-length carbon chains [4–7]. The intense interest in studying the structure and mechanics of carbyne [8–13], besides experimental efforts for fabricating carbyne chains with different lengths is because of its novel electron transport mechanism, possible applications as graphene interconnectors [14], its prospect of being used as a component in atomistic scale circuits [9, 10], and also other possible technological breakthroughs.
The mechanical properties of carbyne have been extensively investigated using various methods including molecular dynamics (MD) simulations, and first-principles calculations [8–10]. However, it is largely unknown how the mechanical properties of this structure are affected by the environmental factors in applications (e.g., filtration and water purification devices), particularly the effect of chain length, temperature and chemical environment on the fracture force of carbyne chains. Recent achievements in studying and synthesizing complex molecular mechanisms based on carbyne chains [15–18] and potential unique applications for these new structures motivated us to investigate those effects. For example, one major application by understanding the mechanics of carbyne is accessing the mechanical strength of graphyne in various working conditions [15, 19]. Graphyne is a two-dimensional carbon allotrope which features similar geometry of graphene by replacing a portion of carbon–carbon bonds in graphene with carbyne chains of certain length. Different carbyne lengths and stacking arrangements can be used to synthesize a variety of structures including α-graphyne, β-graphyne, γ-graphyne and graphdiyne [15, 18, 19].
It has been recently shown that the various graphyne structures can be utilized effectively for various filtration and purification applications, including water purification [15, 20–22]. The supreme efficiency of graphyne structures for water purification applications originate from several unique properties including the small thickness which maximizes permeability, mechanical robustness, and the uniform distribution pores in the membrane [21]. The separation performance of graphyne is strongly affected by the size and resilience of pores, and designing an efficient purification device for separating different contaminants from water needs introducing structures with various pore shapes and sizes. The request of pore geometry can be achieved by using graphyne featuring different carbyne chains in lengths while leaving the mechanics aspect largely unknown. Our results shed light into the mechanics of carbyne chains with different lengths for the first time. We also study the effect of temperature on the mechanical properties of carbyne. In both cases, besides the MD simulations we present a theoretical framework to give a fast and insightful explanation of the carbyne strength at any temperature and chain length. This framework can be used for designing and optimizing an appropriate structure for purification applications. We also investigate the effect of water on the strength of carbyne chains, specifically accounting for the interaction with the surrounding water, and we show here how the rupture force is affected by this interaction.
2. Methods
We focus on the dependency of carbyne chains nanomechanical properties, particularly the strength, to the chain length, temperature and also interaction with water molecules. Two different methods are used in this section; molecular dynamics with the first-principles based ReaxFF force field [23, 24] and a theoretical framework based on the extended Bell model [25, 26]. The presented results shed light on the nanomechanical properties of carbyne chains, and can be used for designing network structures for applications that requires specific porosity and strength, such as graphyne and graphdiyne for water purification.
2.1. Molecular dynamics study
Carbyne chains with different lengths are constructed. In all the chains, both the ends contain a triple carbon–carbon bond and the end carbons are bonded to a hydrogen atom. One of the end carbons is fixed and the other end is displaced to model the uniaxial tension. MD simulations are carried out under a microcanonical (NVT) ensemble (temperature control by a Berendsen thermostat [27], with a time step of 0.1 fs in LAMMPS [28]. The strain rate effect is studied by applying the displacement at different rates and the rate of 1E − 6 fs−1 is selected after making sure the convergence is achieved. For preventing the ambiguities in assuming appropriate cross section for the carbon chain, we report the axial force calculated from the reaction force at the clamped edge instead of using a virial stress. We consider four different temperatures T = 10, 100, 300 and 400 K. All the structures are energy minimized and equilibrated at the specified temperature before applying the mechanical load. For calculating the rupture force of carbyne chains, several simulations are performed for each length at different temperatures and the results are averaged. For studying the effect of water on the fracture force, the carbyne chain is modeled inside a periodic box filled with water molecules and equilibrated for 20 ps before applying the mechanical load on the chain.
2.2. Theoretical model
We model the carbyne chain by N carbon atoms connected by alternating single and triple CC bonds. The chain is subjected to tensile force F before rupture and the probabilities for rupture at the single and triple CC bonds are given by

and
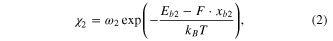
respectively, where  and
and  are the energies of single and triple CC bonds,
are the energies of single and triple CC bonds,  and
and  are the natural vibration frequencies of single and triple CC bonds, and
are the natural vibration frequencies of single and triple CC bonds, and  and
and  are the separation distances of single and triple CC bonds, respectively. The off rate is given by
are the separation distances of single and triple CC bonds, respectively. The off rate is given by
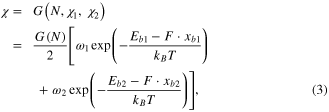
where N is the total number of both single and triple bonds and the total chain length equals to L = 1.38N Å (1.38 is the average of single and triple bond lengths which are 1.54 and 1.22 Å), and  is a function that accounts for the length effect of the chain, subjecting to the condition that
is a function that accounts for the length effect of the chain, subjecting to the condition that  . To be general we take
. To be general we take  as the extended version of Bell model, where a is the power factor and its larger value means the length effect is more significant, as, for example, a = 0 means no length effect and a = 1 means each CC bond behave independently and makes the model converge back to the classical Bell model. Given the loading is almost in constant rate as
as the extended version of Bell model, where a is the power factor and its larger value means the length effect is more significant, as, for example, a = 0 means no length effect and a = 1 means each CC bond behave independently and makes the model converge back to the classical Bell model. Given the loading is almost in constant rate as  , the rupture force as a function of the loading rate and chain length is given by
, the rupture force as a function of the loading rate and chain length is given by
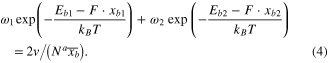
To determine the vibration frequency, we calculate the stiffness of the single and triple CC bond with GAMESS. We use DFT calculation with 6–311G* basis set and BLYP exchange-correlation functional to calculate the CC bond energy as function of distance. For a single CC bond we take the C2H6 and for a triple CC bond we take C2H2 for calculations, and obtain the result as shown in figure 1.
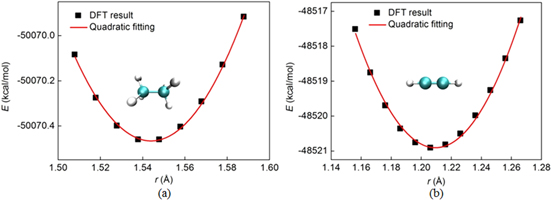
Figure 1. DFT calculation for the system energy as functions of the C-C distance near the equilibrium point that accounts for the bond stiffness of (a) a single bond and (b) a triple bond. The continue curve in each of those subplots is the result of the quadratic fitting according to the DFT calculations, from which the stiffness of  kcal mol−1 Å−2 is measured for single CC bond and 1157.36 kcal mol−1 Å−2 is measured for triple CC bond.
kcal mol−1 Å−2 is measured for single CC bond and 1157.36 kcal mol−1 Å−2 is measured for triple CC bond.
Download figure:
Standard image High-resolution imageBy fitting the result with quadratic functions near the equilibrium point, the spring stiffness is obtained as  kcal mol−1 Å−2 and
kcal mol−1 Å−2 and  kcal mol−1 Å−2. The frequency of the CC bond is calculated via
kcal mol−1 Å−2. The frequency of the CC bond is calculated via
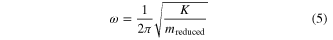
Where  is the reduce mass of the two bonded carbon atoms with
is the reduce mass of the two bonded carbon atoms with  for the mass of each carbon atom. The nature frequencies are measured as
for the mass of each carbon atom. The nature frequencies are measured as  Hz and
Hz and  Hz.
Hz.  and
and  are the bond rupture length which needs to be fitted according to the MD simulation result.
are the bond rupture length which needs to be fitted according to the MD simulation result.
Considering that  ,
,  can be obtained. Thus equation (4) can be simplified to
can be obtained. Thus equation (4) can be simplified to
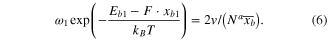
Thereby, equation (6) can be solved to obtain
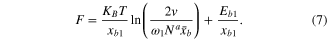
We use the MD simulation results in the following section to calibrate the parameters and use this equation to predict the dependence of rupture force to the chain length and temperature.
3. Results and discussion
Uniaxial tension tests are carried out for carbyne chains of 7–60 Å length at temperature range of 10–400 K. The force-extension curves for two chains of 30 and 60 Å at different temperatures are shown in figure 2. As shown in this figure, the temperature change slightly affects the force extension curves in short carbyne chain, while it has a more prominent effect on the mechanical response of longer chains. At higher temperatures, the force-extension curved in long chains reveals larger fluctuations caused by the vibration of the chain during loading.
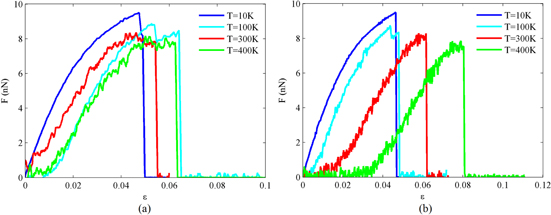
Figure 2. Force-extension response of carbyne chains with (a) 30 Å and (b) 60 Å at different temperatures.
Download figure:
Standard image High-resolution imageThe change of fracture force as a function of chain length is shown in figure 3(a) for varied temperatures. The MD simulations are shown with separate points and the error bars are calculated based on several simulations and also by considering the fluctuations in the force value at the fracture point. The rupture force as a function of temperature for chains with different lengths is also shown in figure 3(b).
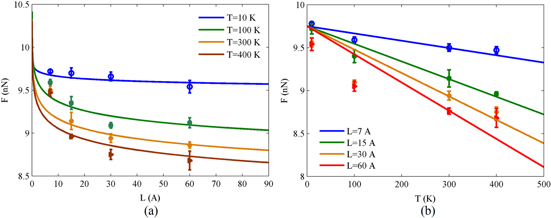
Figure 3. Carbyne chain rupture force (a) as a function of chain length at different temperatures and (b) as a function of temperature for chains with different lengths. The data points show the MD simulation and continuous lines correspond to the theoretical results (see equation (7)).
Download figure:
Standard image High-resolution imageThe continuous lines in these figures show the results obtained by the theoretical framework of section 2.2. For obtaining the theoretical results we have used the numerical values of  Hz, L = 1.38N Å and
Hz, L = 1.38N Å and  m s−1 as what is used in the MD simulations. We fit the data points of the chain strength as the function of T and N by using equation (7) and obtain the numerical values of
m s−1 as what is used in the MD simulations. We fit the data points of the chain strength as the function of T and N by using equation (7) and obtain the numerical values of  ,
,  ,
,  and
and  as summarized in tables 1 and 2.
as summarized in tables 1 and 2.
Table 1.
The parameters  ,
,  ,
,  and
and  at different temperatures.
at different temperatures.
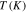
|
|
|
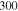
|
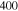
|
|---|---|---|---|---|
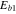 (kcal mol−1) (kcal mol−1) |
56.2 | |||
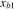 (Å) (Å) |
0.4 | |||
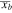 (Å) (Å) |
0.01 | |||

|
11.76 | 4.36 | 1.60 | 1.30 |
Table 2.
The parameters  ,
,  ,
,  and
and  at different chain lengths.
at different chain lengths.
| L(Å) | 7 | 15 | 30 | 60 |
|---|---|---|---|---|
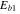 (kcal mol−1) (kcal mol−1) |
56.2 | |||
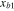 (Å) (Å) |
0.4 | |||
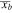 (Å) (Å) |
0.01 | |||

|
0.05 | 1.46 | 1.76 | 1.86 |
Figure 3(a) shows that the logarithmic decay of the fracture force by increasing the chain length is predicted by the theoretical framework with an acceptable accuracy compared to the MD simulations. Also, it is clearly shown that the rupture force decreases by increasing the temperature for all the selected lengths. The linear decrease of rupture force by increasing the temperature is shown in figure 3(b) which is observed in both the theoretical and numerical results.
The change of parameter a in the theoretical model can be related to the bond order distribution in the carbyne chains with different lengths. The bond order at different lengths is studied in [9] and it is shown that for the shorter chains the bond order of the carbon atom at the location of failure remains higher at larger strain values. This phenomenon can be explained by considering the fact that in a short chain the carbon atoms can relax over a smaller distance, and thereby the chain is not completely broken after reaching the maximum stress [9]. This effect is reflected in the change of parameter a in our theoretical model. For the short chains this parameter is chosen smaller, and for the larger lengths by increasing the length effect (which is also reflected in the change of bond orders), this parameter increases accordingly. The change of this parameter at different temperatures reflects the interaction between temperature and length effects in the chain. At lower temperatures, in absence of local fluctuations, the length effect is more significant and a larger value for a is selected, while at higher temperature local fluctuations overcome the length effects and smaller a is selected to match the theoretical model with the MD results by performing a least square curve fitting between the theoretical and MD results.
In order to study the effect of water molecules on the rupture force, carbyne chains are modeled inside water-filled periodic boxes as shown in figure 4(a). The system is equilibrated for 40 ps under isothermal–isobaric ensemble (NPT) at T = 300 K and zero pressure. Two different lengths of 7 and 15 Å are studied. During the equilibration stage, the carbyne chain deforms under the interaction of water molecules as shown in figure 4(b). One of the chain ends is fixed and the other end is displaced with a constant strain rate for modeling the uniaxial loading. The deformation of short and long carbyne chains up to the breaking point is shown in figure 4(b). The effect of water molecules on the deformed shape is obvious in all the loading steps. This effect elucidates the necessity of considering such simulations in designing carbyne-based structures, particularly graphyne, for applications in which the structure interacts with water, i.e. the purification devices. Interaction with the water environment also affects the force-extension response and the rupture force of carbyne chains as shown in figure 5. The very initial stages of unfolding with almost zero force in the force-extension graphs are not shown in these figures.
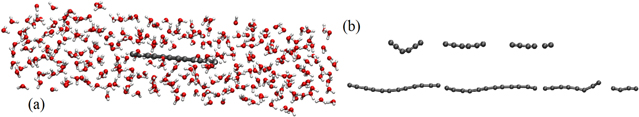
Figure 4. (a) Carbyne chain (of 15 Å in length) inside the water-filled box before equilibration and (b) unfolding and stretching carbyne chains surrounded by water molecules with 7 (upper) and 15 Å (lower) length. Water molecules in (b) are not shown for clarity.
Download figure:
Standard image High-resolution image
Figure 5. The force extension of carbyne chains with (a) 7 Å and (b) 15 Å length. The force extension curve is compared for the original chains and carbyne chains in water.
Download figure:
Standard image High-resolution imageAs shown in figure 5, the water molecules slightly affect the response of both carbyne chains. The force-extension of chains in water is associated with several regions with different slopes. Studying the deformation of the chain shows that the deformation mechanism in water consists of unbending the folded chain and extension of the bonds in the chain. The deformation mechanism switches between unbending and stretching due to the varying interaction of the chain with the surrounding water molecules.
Both the rupture force and ultimate strain of the chains are slightly improved in water. This phenomenon can be explained by studying the bond orders in the chain during the mechanical loading. The bond order of the carbon atom at the failure location is shown in figure 6 for the carbyne chains with 7 and 15 Å length (the broken bond is always a single bond since it is weaker compared to the triple carbon–carbon bond).

Figure 6. Bond order of the failed single carbon–carbon bond in carbyne chains with (a) 7 Å and (b) 15 Å length with and without the interaction of water molecules.
Download figure:
Standard image High-resolution imageWe find that the interaction with water molecules improves the bond stability, which is reflected at the higher values of bond order. Also, for the chains in water, a residual covalent bonding is observed at higher strain values, which explains the larger deformation of carbyne chain before breaking.
In a variety of applications, including the water purification structures [15, 20], carbyne may be operating in either acidic or basic solutions. We examine the effect of these chemical environments on the mechanical properties of carbyne chains in the following. In order to simulate the basic chemical environment, we replace a portion of water molecules in the simulation box with hydroxide groups  , and the acidic solution is simulated by replacing some of the water molecules with carbonic acid
, and the acidic solution is simulated by replacing some of the water molecules with carbonic acid  . These acidic and basic solutions are selected by considering the available elements in the ReaxFF force field used in this paper. In order to study the extreme conditions of acid or base solutions, high concentrations of hydroxide and carbonic acid are selected (∼20% of total water molecules). However, further simulations with a lower concentration of these solutions show that the general trend is not strongly affected by the concentration if the system is equilibrated for a longer time or enough ions are near the carbyne chain at the beginning of simulation.
. These acidic and basic solutions are selected by considering the available elements in the ReaxFF force field used in this paper. In order to study the extreme conditions of acid or base solutions, high concentrations of hydroxide and carbonic acid are selected (∼20% of total water molecules). However, further simulations with a lower concentration of these solutions show that the general trend is not strongly affected by the concentration if the system is equilibrated for a longer time or enough ions are near the carbyne chain at the beginning of simulation.
Figures 7(a) and (b) compare the force extension curves for 7 and 15 Å pristine carbyne chains with the carbyne chains is acidic and basic solutions. It is observed that the basic environment decreases the chain strength, while the strength in acidic solution is slightly increased. Further analyzing the mechanical response of carbyne chains in the basic solution shows that the force-extension curves has two consequent peaks before the final rupture of the chain. In order to better understand this phenomenon, we study the chemical reactions around the carbyne chain in basic solution as shown in figure 8. The upper row in figure 8 shows three snapshots of the carbyne chain with 15 Å length in the basic solution during axial loading. As it is shown, when the carbyne chain ruptures, an oxygen atom in a near  group loses its hydrogen atom and bonds with the carbon atoms in the carbyne chain at the ruptured ends to form a CCOCC chain. Breaking the chain and splicing the oxygen into the carbon chain corresponds to the first pick in the force-extension curve in figure 7. By further loading the healed carbyne chain, the new bond breaks at the second peak in the force-extension curve. A detailed view of the reaction for smaller carbyne chain is shown in lower rows in figure 8, in which only the carbyne chain and the
group loses its hydrogen atom and bonds with the carbon atoms in the carbyne chain at the ruptured ends to form a CCOCC chain. Breaking the chain and splicing the oxygen into the carbon chain corresponds to the first pick in the force-extension curve in figure 7. By further loading the healed carbyne chain, the new bond breaks at the second peak in the force-extension curve. A detailed view of the reaction for smaller carbyne chain is shown in lower rows in figure 8, in which only the carbyne chain and the  group that forms bonds with the ruptured ends are shown. The process shows how that hydroxide group is attracted to carbine end during the equilibration and how the oxygen atoms fills the gap in the carbyne chain after breaking.
group that forms bonds with the ruptured ends are shown. The process shows how that hydroxide group is attracted to carbine end during the equilibration and how the oxygen atoms fills the gap in the carbyne chain after breaking.

Figure 7. The force extension of carbyne chains with (a) 7 Å and (b) 15 Å length. The force extension curve is compared for the pristine chains and carbyne chains in acidic and basic solutions.
Download figure:
Standard image High-resolution image
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. The rupture of carbyne chains with (a) 15 Å and (b) 7 Å length in a basic solution. The snapshots show the steps of healing the broken chain by an oxygen atom from the OH− group attracted to the chain end during equilibration. Water molecules in (b) are not shown for clarity.
Download figure:
Standard image High-resolution image{kind=link}
4. Conclusion
In this work the effect of various parameters, including the chain length, temperature and operating environment, on the mechanical properties of carbyne were studied. Carbyne and carbyne-based structures are supreme candidates for several applications, i.e. water purification, and designing such materials and devices requires a better understanding of carbyne mechanical properties. Molecular dynamics (MD) simulations are performed for calculating the rupture force of carbyne chains with different lengths at different temperatures. In order to facilitate the design procedure, a theoretical framework is presented that is calibrated based on the MD simulations for assessing the rupture force. The effect of water molecules' interaction and the acidic and basic solutions are also studied on the mechanical properties, which presents fundamental knowledge for studying carbyne-based filtration and purification devices.
Acknowledgements
The authors gratefully acknowledge the support from ARO through a MURI award, No. W911NF-09–1-0541.