
Abstract
A density functional theory study of the BiS2 superconductors containing rare-earths: LnO1−xFxBiS2 (Ln = La, Ce, Pr, and Nd) is presented. We find that CeO0.5F0.5BiS2 has competing ferromagnetic and weak antiferromagnetic tendencies, the first one corresponding to experimental results. We show that PrO0.5F0.5BiS2 has a strong tendency for magnetic order, which can be ferromagnetic or antiferromagnetic depending on subtle differences in 4f orbital occupations. We demonstrate that NdO0.5F0.5BiS2 has a stable magnetic ground state with weak tendency to order. Finally, we show that the change of rare earth does not affect the Fermi surface, and predict that CeOBiS2 should display a pressure induced phase transition to a metallic, if not superconducting, phase under pressure.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The excitement generated by the recent finding of two new superconductors, Bi3O2S3 and LaO1−xFxBiS2 [1, 2], led to the unveiling of a whole class of superconductors containing BiS2 bilayers. These compounds share a common two-dimensional structure with alternating BiS2 bilayers and spacer layers. It is the BiS planes in the BiS2 bilayers which are thought to be responsible for superconductivity in these compounds [3]. Soon after the discovery of superconductivity in LaO1−xFxBiS2, chemical substitution to tune the properties was attempted. The first research axis was to replace the lanthanum atom by other lanthanides: cerium, praseodymium, neodymium, europium and ytterbium [4–12]. Hole and electron doping of the parent compound has also been studied in SrxLa1−xFBiS2 [13] and La1−x MxOBiS2 (M = Ti, Zr, Hf, Th) [14, 15] respectively.
The electronic and thermodynamic properties of this group of materials are indeed interesting and theoretical suggestions for the mechanism of superconductivity range from spin-fluctuation mediated superconductivity [16], to proximity to ferroelectricity and charge density wave (CDW) instabilities [17, 18]. Recent work even revealed the potential of related materials for photovoltaic applications [19]. Ferromagnetism has been shown experimentally to coexist with superconductivity in CeO0.5F0.5BiS2 [7, 20–27], and quantum critical fluctuations of the magnetic moments have been observed in CeOBiS2 [28].
The electronic structure of the two first compounds to be found, Bi3O2S3 [3] and La(O,F)BiS2 [17, 29–31], was calculated. These calculations indicated that the superconducting electrons are a mixture of Bismuth 6 px, y and Sulphur 3 px, y states [29, 30]. These form eight bands, four of which are under the Fermi level, when the other four either are above the Fermi level or cross it.
The coupling mechanism for superconductivity has been investigated in various ways. Electron–phonon interactions have been calculated in La(O,F)BiS2, and yield a large electron–phonon coupling constant, suggesting superconductivity in this compound is strongly coupled and conventional [17, 18, 32]. Renormalisation-group calculations suggested triplet pairing and weak topological superconductivity [33, 34], a possibility studied in the context of quasiparticle interference [35]. Random phase approximation was applied to a two-orbital model [30], leading to an extended s-wave or d-wave pairing [16, 36].
In this paper we present our findings from electronic structure calculations for the stoichiometric and doped series of materials LnO1−xFxBiS2 with Ln = La, Ce, Pr and Nd, and x = 0, 0.5. The doped phases calculated have been reported as being superconducting, and the parent phases have been reported as being non-superconducting.
We first show that the change of rare earth does not affect the Fermi surface and that the conduction electrons are confined to the bismuth-sulphur planes. We then demonstrate that CeO0.5F0.5BiS2 has competing ferromagnetic and weak antiferromagnetic tendencies. The ferromagnetic tendency is in agreement with experimental results. We show that PrO0.5F0.5BiS2 has a strong tendency to magnetic order, which could be ferromagnetic or antiferromagnetic depending on subtle differences in 4f orbital occupations. We show that NdO0.5F0.5BiS2 has a stable weakly antiferromagnetic ground state. Finally, we predict that applying pressure to CeOBiS2 would make it metallic, and possibly superconducting.
2. Methods
We limit our study to materials which crystal structure has been experimentally determined, with the exception of the Ce doped compound. We have been able to provide a structure through relaxation methods for the Ce compound, since the availability of the La, Pr and Nd compounds provides a reasonable reliability on the starting point.
Band structures and densities of states were calculated within density functional theory using the SIESTA method [38, 39], implementing the generalized gradient approximation (GGA) in the shape of the Perdew, Burke, and Ernzerhof functional [40] and the GGA+U method. It uses norm-conserving pseudopotentials to replace the core electrons, while the valence electrons are described using atomic-like orbitals as basis states at the double zeta polarized level. These pseudopotentials include scalar relativistic corrections. Spin–orbit coupling was not included but has been shown in previous work to be of no relevance to the main features at the Fermi level in this family of materials [3, 18]. Details on the basis sets used are available in the supplemental material (stacks.iop.org/JPhysCM/28/345504/mmedia).
Calculations for x = 0.5 have been performed for all the compounds. All these structures share the same space group: P4/nmm (figure 1). The calculations for the lanthanum compound were performed without spin polarisation. However for Ln  La, the presence of unpaired 4f electrons demands the use of spin polarisation in the calculations. Calculations for the La compound were performed using experimental data for the geometry obtained after high pressure treatment of the sample, for which the maximal superconducting Tc (10.6 K) was found [1]. For the Pr and Nd compounds, we used structural experimental data for the systems for which Tc of 5.5 K [5] and 5 K were found [6] respectively.
La, the presence of unpaired 4f electrons demands the use of spin polarisation in the calculations. Calculations for the La compound were performed using experimental data for the geometry obtained after high pressure treatment of the sample, for which the maximal superconducting Tc (10.6 K) was found [1]. For the Pr and Nd compounds, we used structural experimental data for the systems for which Tc of 5.5 K [5] and 5 K were found [6] respectively.

Figure 1. Crystal structure of LaOBiS2, represented in VESTA [37]. All the compounds studied share this same structure, with replacement of lanthanum by other lanthanides, or of oxygen by fluorine. The black solid line represents the unit cell, which contains two formula units.
Download figure:
Standard image High-resolution imageNo atomic coordinates have been reported for the Ce compound yet, so we obtained a geometry from density functional theory (DFT) by relaxing the structure (both atoms and cell), starting from the structure known for the La compound. Two relaxations were performed, one with a k-grid of  points, a standard set of parameters and without U, and one with a k-grid of
points, a standard set of parameters and without U, and one with a k-grid of  points, a high quality set of parameters and with +U. Both relaxations were performed in the ferromagnetic configuration. The comparison of the results shows the relaxation is well converged: the lattice constants differ by approximately 2 mÅ, and the fractional atomic coordinates by less than 0.0004. The details of the crystal structures are available in the supplementary material. The obtained atomic positions and a lattice parameter are very close to the ones of the Pr doped compound. The c lattice parameter is significantly smaller than the ones of the other doped compounds. This is in accordance with mismatches between DFT and experiments in the inter-layer forces noted in other compounds of this family of superconductors [3].
points, a high quality set of parameters and with +U. Both relaxations were performed in the ferromagnetic configuration. The comparison of the results shows the relaxation is well converged: the lattice constants differ by approximately 2 mÅ, and the fractional atomic coordinates by less than 0.0004. The details of the crystal structures are available in the supplementary material. The obtained atomic positions and a lattice parameter are very close to the ones of the Pr doped compound. The c lattice parameter is significantly smaller than the ones of the other doped compounds. This is in accordance with mismatches between DFT and experiments in the inter-layer forces noted in other compounds of this family of superconductors [3].
There are two O/F sites in each unit cell of every doped compound. It has been shown that disorder in the O and F positions has a very minor effect on the electronic structure at the level discussed in this work [41]. As already done in [17, 29, 31], we assign O to (0,0,0) and F to (0.5,0.5,0) in the La doped compound and O to (0.75,0.25,0) and F to (0.25,0.75,0) in the other doped compounds. This changes the space group of the crystal structures of the doped compounds to P m2.
m2.
We also performed calculations on two parent phases: LaOBiS2 and CeOBiS2. Their space group is also P4/nmm. We used experimental structural data for both [42, 43], and performed spin-polarised calculations in the ferromagnetic configuration for CeOBiS2. There are no experimental data available for the other two parent compounds to our knowledge.
We performed GGA + U calculations [44] for all the compounds except the lanthanum ones, because of the known strong correlation of the 4f electrons in cerium, praseodymium and neodymium. The current implementation of DFT + U in SIESTA is based on the formulation by Dudarev et al [45] which combines the two parameters U and J to produce an effective Coulomb repulsion  which we call U in the following. The double-counting scheme used is also the one described by Dudarev et al [45]. Given the intrinsic difficulty in the ab initio determination of a value of U [46], we explored the behaviour of the system for U varying from 3 to 7 eV. The influence of U on relaxations was investigated and proven to be negligible. Here we discuss results obtained with U = 5 eV, given that this value is standard for the atoms we are dealing with [47, 48]. We tested the robustness of the results for U between 3 eV and 7 eV.
which we call U in the following. The double-counting scheme used is also the one described by Dudarev et al [45]. Given the intrinsic difficulty in the ab initio determination of a value of U [46], we explored the behaviour of the system for U varying from 3 to 7 eV. The influence of U on relaxations was investigated and proven to be negligible. Here we discuss results obtained with U = 5 eV, given that this value is standard for the atoms we are dealing with [47, 48]. We tested the robustness of the results for U between 3 eV and 7 eV.
Monkhorst–Pack sampling of the Brillouin zone was used for all calculations. k-point sampling proved to be quite delicate, and large sets were used to obtain reasonably converged results, especially for the convergence of magnetic results. The grids used were  for the Ce and Nd compounds,
for the Ce and Nd compounds,  for the Pr compound and
for the Pr compound and  for the La compound. The band structure results converged for lower sets, and the results in figure 6 were obtained for
for the La compound. The band structure results converged for lower sets, and the results in figure 6 were obtained for  sets.
sets.
3. Results
3.1. Band structures
We calculated the band structure of the four doped compounds. We observe many similarities between them, due to their proximity in terms of composition and crystal structure (figure 2). More precisely, we find the same four bands (or eight bands with spin polarisation) near the R and X points, and two (or four) bands near the middle of the A–Z segment close to the Fermi level in all the compounds. They are similar to the ones in the Bi-O-S compounds [3]. They are crossing the Fermi level for the doped compounds, and lie above the Fermi level for the two parent phases. The projected densities of states indicate that these bands are formed from Bi and S states. It has already been shown in Bi3O2S3 and in the La compound that the main contribution to these bands comes from Bi 6p states and S 3p states. It is confirmed in all the systems considered here, as figure 3 shows. There we plotted the results for CeO0.5F0.5BiS2, these being similar in all the compounds considered. The position of the Fermi level with respect to the bottom of these bands is very similar in all the compounds with x = 0.5. The main effect of the fluorine doping is a shift of the Fermi level, as reported previously [29–31].

Figure 2. Band structures (left) and densities of states projected onto the basis orbitals for Bi (red), S (green), Ln (blue), O (lilac), and F (light blue) (right) for the four doped compounds. For the three compositions having 4f electrons, we use U = 5. The lanthanum compound band structure is non spin polarised, whereas the other three are spin polarised, in the ferromagnetic configuration.
Download figure:
Standard image High-resolution image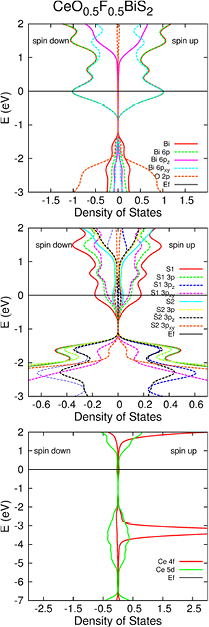
Figure 3. Densities of states in CeO0.5F0.5BiS2, projected onto several basis orbitals. S1 stands for the in-plane sulphur atom, and S2 stands for the out-of-plane sulphur atom. The label pxy is a shorthand for px and py orbitals which have the same density of states. We use U = 5 and spin polarisation.
Download figure:
Standard image High-resolution imageTo explore further the character of the bands just below the Fermi level, we plotted the local density of states (LDOS) resolved in real space for the x = 0.5 compounds for energies integrated between −0.5 eV and 0 eV (figure 4). The results confirm what had been found in other BiS2-based compound: all these electrons accumulate around the BiS planes [3]. This can also be seen in the partial density of states at the Fermi level (figure 3) which is very low for the out-of-plane sulphur atoms. This raises the question of the existence of superconductors that have BiS planes without BiS2 layers. Some pz participation is involved in the slight puckering of the BiS planes, inducing a clear distortion (along z) of the LDOS.

Figure 4. Local density of states of CeO0.5F0.5BiS2 in real space integrated in the energy range [−0.5,0], plotted with XCrySDen [49]. Half of the atoms represented are not in the plane corresponding to the density plot: the atoms in the unit cell are arranged on two planes parallel to the x-z plane. The density plot corresponds to only one of these two planes. The BiS planes (horizontal and perpendicular to the figure) gather the vast majority of the density.
Download figure:
Standard image High-resolution imageThe lanthanide bands are at least 3 eV below and 0.5 eV above the Fermi level. The Fermi surface is robust to lanthanide substitution and to the change of U between 3 eV and 7 eV. There is no difference in the density of states at the Fermi energy between compounds, the lanthanides' bands being far from the Fermi level, as expected, because of the strong correlation of the 4f electrons. The extra electrons remain in the lanthanide bands and away from the Fermi level, hence the change of lanthanide has almost no effect. The Fermi surface is almost identical in the four cases, and does not seem to be responsible for the substantial change observed in Tc, nor does the band structure as a whole.
3.2. Magnetic order
We considered the magnetic properties of the doped compounds. They each have two Ln atoms per unit cell. We compared the total energies of the ferromagnetic configuration, where the spins of the two atoms are aligned, and of the only antiferromagnetic configuration available in one unit cell, where the spins of the two atoms are antialigned.
Bulk cerium, praseodymium and neodymium have respectively one, three and four 4f electrons per atom. The two-electrons difference between Ce and Pr is due to the fact that bulk cerium has one electron in a 5d band, whereas bulk praseodymium and neodymium have not. However when we place these elements in the compounds considered here, each atom of these species has one 5d electron, thereby restoring the sequence. As expected the 4f electrons are polarised and the 5d are not. The spin polarisation  is therefore 1, 2 and 3 electrons for the Ce, Pr and Nd atoms in their respective compounds. The 5d electron is unpolarised and strongly hybridised with other bands (figure 3).
is therefore 1, 2 and 3 electrons for the Ce, Pr and Nd atoms in their respective compounds. The 5d electron is unpolarised and strongly hybridised with other bands (figure 3).
These compounds share the space group P4/nmm. The lanthanide atoms are on the sites (1/4,1/4,z) and (3/4,3/4,− z). These have a 4 mm symmetry. Hence the sites have the point group C4v. The nearest neighbours of the lanthanide atoms are 4 oxygen atoms. As a way of characterising the 4f orbitals in this environment, we treat the Ln and the 4 O atoms as a molecule and look for the symmetries of the molecular orbitals for bonding in that molecule. Finally, we obtain that all the 4f orbitals are allowed by symmetry to hybridise with the oxygen atoms, except the 4 orbital, but it is never occupied, see below.
orbital, but it is never occupied, see below.
3.2.1. CeO0.5F0.5BiS2.
In the cerium compound we find two different competing states which are quasi-degenerate in energy. It does not make sense for us to try to establish which one is favoured since either state appears in the calculations under very slight changes of the technical parameters, even for very tightly converged settings (see the methods section).
In the ferromagnetic configuration, calculations always converge on the same state which we call state F. But in the antiferromagnetic configuration, calculations converge on two different states, state AF1 and state AF2. They differ by the 4f orbital chosen by the 4f electrons in the Ce atoms. The orbital occupations of the 4f electron are summarised in table 1. The total energy of state AF2 is higher than the one of state F, while the total energy of state AF1 is lower than the one of state F (table 2). Therefore we obtain two competing states, F and AF1.
Table 1. Orbitals occupied by the 4f electron in each cerium atom in the unit cell of CeO0.5F0.5BiS2 in the two antiferromagnetic configurations AF1 and AF2.
| Configuration | 4f occupations | |
|---|---|---|
| 1st Ce atom | 2nd Ce atom | |
| F | 4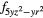 |
4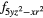 |
| AF1 | 4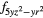 |
4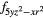 |
| AF2 | 4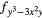 |
4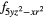 |
Note: The labelling order of the cerium atoms is arbitrary.
Table 2. Total energies of the various magnetic configurations obtained for CeO0.5F0.5BiS2 with respect to the energy of F.
| Configuration | Total energy (eV) |
|---|---|
| F | 0 |
| AF1 | −0.0124 |
| AF2 | 0.1105 |
In state AF1 the spin–spin interaction is weak, with a slight tendency for them to order antiferromagnetically, as manifested by the energy difference with respect to ferromagnetic order of 12 meV per cell. In state F the ferromagnetic order is more substantially favoured, by 111 meV per cell.
The band structures and density of states for states AF1 and AF2 are plotted in figure 5. The bands are very similar, and quite indistinguishable around the Fermi level. The only difference is that in state AF2 the bands around the peak in 4f density of states are slightly higher than in state AF1. However, the projected density of states on the different f-orbitals clearly show the change of orbital character of the 4f electron.
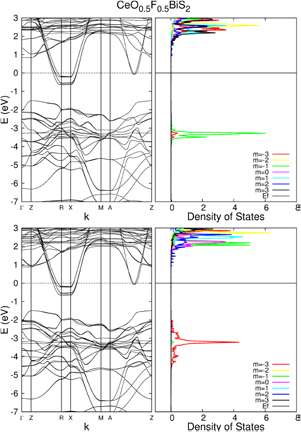
Figure 5. Band structures and density of 4f states of CeO0.5F0.5BiS2 in the antiferromagnetic configuration, in the two states that it converges to. The projected densities of states were plotted using a peak width of 0.01 eV, smaller than the standard 0.2 eV used in the rest of this work. State AF1 is at the top, and state AF2 at the bottom. The decomposition of density of states on the 9 orbitals clearly shows that the two configurations differ by the jump of one electron from one orbital to another.
Download figure:
Standard image High-resolution imageThe ferromagnetic state is compatible with experimental observations of ferromagnetism in this compound at low temperature. The presence of the weak antiferromagnetic state suggests that this system may be close to an antiferromagnetic instability associated to a change of orbital for the 4f electron, which would suggest possibly interesting phenomena associated with this coupling between spin ordering and choice of orbital.
3.2.2. PrO0.5F0.5BiS2.
A similar instability is observed in the Pr compound, in which each praseodymium atom has two electrons in the 4f shell. We obtain an effective degeneracy between two states for both the ferromagnetic (states F1 and F2) and the antiferromagnetic (states AF1 and AF2) configurations. In all cases the spin polarisation is 2 electrons on each atom. The two states on which each configuration converges only differ by the orbital occupations of the 4f electrons of the praseodymium atoms. The orbital occupations of the 4f electron are summarised in table 3.
Table 3. Orbitals occupied by the 4f electron in each praseodymium atom in the unit cell of PrO0.5F0.5BiS2 in the four configurations FM1, FM2, AF1 and AF2.
| Configuration | 4f occupations | |||
|---|---|---|---|---|
| 1st Pr atom | 2nd Pr atom | |||
| 1st electron | 2nd electron | 1st electron | 2nd electron | |
| F1 | 4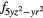 |
4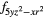 and 4 and 4 |
4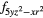 |
4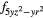 and 4 and 4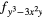 |
| F2 | 4fxyz | 4 and 4 and 4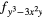 |
4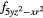 |
4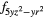 and 4 and 4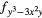 |
| AF1 | 4fxyz | 4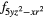 and 4 and 4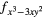 |
4fxyz | 4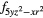 and 4 and 4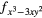 |
| AF2 | 4fxyz | 4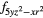 and 4 and 4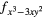 |
4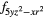 |
4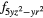 and 4 and 4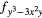 |
Note: The labelling order of the praseodymium atoms and electrons is arbitrary.
For the same input parameters we obtain either F1 and AF1 or F2 and AF2. Therefore for a given set of parameters we obtain two energy differences, AF1-F1 and AF2-F2. These are 92.9 meV in favor of ferromagnetism and 94.8 meV in favor of antiferromagnetism, hence we obtain two competing states, AF2 and F1 (table 4).
Table 4. Total energies of the various magnetic configurations obtained for PrO0.5F0.5BiS2 with respect to the total energy of F1.
| Configuration | Total energy (eV) |
|---|---|
| F1 | 0 |
| F2 | 0.094527 |
| AF1 | 0.092904 |
| AF2 | −0.000248 |
The Pr compound has therefore two competing states, with very close energies. Both display magnetic ordering, with a strength very close to the one in CeO0.5F0.5BiS2, one ferromagnetic and the other antiferromagnetic.
3.2.3. NdO0.5F0.5BiS2.
In this case we find a single ferromagnetic state and a single antiferromagnetic one. We obtain a difference between the two spin states of 3.3 meV favouring antiferromagnetic order. This is very small, indeed it is two orders of magnitude lower than the difference in favor of ferromagnetism in the cerium compound. It is therefore likely that the magnetic order in this material will arise at temperatures much lower than the Curie temperature of the order of 1 K measured in the cerium compound.
The presence of an instability in the cerium and praseodymium doped compounds is consistent with the well known valence fluctuation phenomenon which has been studied in other compounds containing these two atoms [50, 51]. To the knowledge of the authors, no neodymium compound exhibits this phenomenon, which is in line with its absence in NdO0.5F0.5BiS2.
The results discussed above for the three compounds do not change qualitatively with the choice of U value, as shown in figure 6, where the energy difference between the antiferromagnetic and the ferromagnetic configurations are shown for the three compounds. Calculating the total energy of other magnetic configurations using a larger unit cell has been attempted but definite conclusions could not be achieved, partly because of the described instabilities.
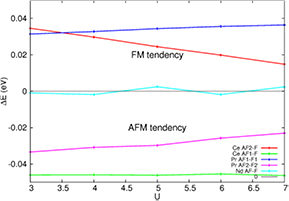
Figure 6. Difference of total energy per unit cell of LnO0.5F0.5BiS2 (Ln = Ce, Pr, Nd) between antiferromagnetic state and ferromagnetic states, for U varying between 3 and 7. The k-point sampling set used here is smaller than in the rest of the text (see the methods section). The key is labelled with names of magnetic configurations defined in the main text. A positive difference means the tendency is ferromagnetic.
Download figure:
Standard image High-resolution image3.3. CeOBiS2 under pressure
Finally, we consider the effect of pressure on CeOBiS2. Without electron doping the system is insulating with the BiS bands clearly above the Fermi level. The question is whether pressure can close that gap. The band structure of CeOBiS2 has been calculated with respect to pressure and U (figures 7 and 8). At ambient pressure, the system is insulating. When pressure increases, the gap closes and some band cross the Fermi level. The transition pressure varies with U, from 1.2 GPa for U = 3 eV to 17.5 GPa for U = 7 eV. The bands crossing the Fermi level are not only the BiS bands: other bands are crossing. These are cerium bands, hybridised with oxygen and sulphur bands. Indeed, the main effect of pressure is to raise the cerium oxide bands which in turn dope the BiS bands. This effect is similar to the stoichiometric doping of the BiS bands by the sulphur dimers bands in Bi3O2S3 [3]. The contribution of the oxygen and sulphur bands decreases with pressure but remains substantial, as can be seen in the density of states. The composition of the BiS bands does not change with pressure, it is always dominated by the same Bi 6p and S 3p states. Finally, the conduction band is narrowed whereas the valence band is slightly widened with pressure. Detailed plots of the band structures and densities of states, and crystal structures obtained are available in the supplementary material.

Figure 7. Pressure / U phase diagram of CeOBiS2.
Download figure:
Standard image High-resolution image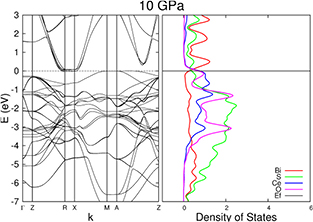
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. Band structure (left) and density of states projected onto the basis orbitals for Bi (red), S (green), Ce (blue), and O (lilac) (right) for CeOBiS2 under 10 GPa. We use U = 5 and spin polarisation.
Download figure:
Standard image High-resolution image{kind=link}
The insulating state at ambient pressure is consistent with the fact that no superconductivity has been observed in this compound. It shows that there should be a insulator-metal transition under pressure, which also could be superconducting at low temperature. The transition pressures obtained have rather large uncertainties given that they have been obtained through DFT-GGA (with its well-known bad gap problem), but we can still conclude that it seems likely the transition temperature could be reachable by commonly known techniques. Of course, this pressure also depends heavily on the U parameter relevant for the compound.
The type of 'doping' involved is different from the fluorine type. Indeed, here pressure brings the valence and the conduction bands together until they cross. Pressure has opposite effects on the valence and conduction bands: the conduction band is narrowed and its composition does not change, whereas the valence band is widened and its hybridisation with oxygen and sulphur orbitals is decreased.
4. Conclusion
We calculated the band structure of the four doped compounds and showed that the change of rare earth does not affect the Fermi surface. We calculated the local density of states, which clearly indicates that the conduction electrons are confined to the bismuth-sulphur planes.
We calculated the difference in energy between the two possible magnetic configurations one can obtain in one unit cell of LnO1−xFxBiS2 (Ln = Ce, Pr, and Nd). We demonstrated that CeO0.5F0.5BiS2 has competing ferromagnetic and weak antiferromagnetic tendencies. The first case corresponds to experimental results. PrO0.5F0.5BiS2 has a strong tendency to order, which could be ferromagnetically or antiferromagnetically depending on subtle differences in 4f orbital occupations. NdO0.5F0.5BiS2 has a stable weakly antiferromagnetic competing state.
Finally, we did predict that applying pressure to CeOBiS2 would make it metallic, and possibly superconducting, through raising the cerium oxide bands. This type of doping of the BiS bands has not been investigated in the BiS2 family so far and could have novel properties.
Acknowledgments
We would like to thank Arman Khojakhmetov for his exploratory work, and Gilbert G Lonzarich, Peter Littlewood, Pablo Aguado Puente, Stephen Rowley, Sebastian Haines, Cheng Liu, Daniel Molnar and Adrien Amigues for fruitful discussions. We acknowledge computing resources of the Spanish Supercomputer Network (RES), and support from EPSRC, Corpus Christi College, and the Winton Programme for the Physics of Sustainability.