
Abstract
The ionization and fragmentation of two selenium containing hydrocarbon molecules, methylselenol (CH3SeH) and ethylselenol (C2H5SeH), by intense (>1017 W cm−2) 5 fs x-ray pulses with photon energies of 1.7 and 2 keV has been studied by means of coincident ion momentum spectroscopy. Measuring charge states and ion kinetic energies, we find signatures of charge redistribution within the molecular environment. Furthermore, by analyzing fragment ion angular correlations, we can determine the laboratory-frame orientation of individual molecules and thus investigate the fragmentation dynamics in the molecular frame. This allows distinguishing protons originating from different molecular sites along with identifying the reaction channels that lead to their emission.
Export citation and abstract BibTeX RIS
1. Introduction
X-ray free-electron lasers (XFELs) such as the Linac Coherent Light Source (LCLS) [1] at the SLAC National Accelerator Laboratory in the USA and SACLA [2] at Spring-8 in Japan provide ultra-short x-ray pulses with unprecedented intensities of 1017 W cm−2 and above. While a number of experimental studies have explored the interaction of these ultra-intense x-ray pulses with isolated atoms [3–8] and small molecules [9–15], as well as with nano-scale rare gas clusters [16–18], only a few results are published to date on intermediate polyatomic systems [19].
In our previous study of the x-ray multi-photon ionization of methylselenol molecules [19], we found that after localized inner-shell multiple ionization of the selenium atom, the created charges are rapidly distributed over the entire molecule. The resulting ultra-fast Coulomb explosion leads to significant changes of the bond length already on the time scale of 5 to 10 fs, defined by the x-ray pulse length and the characteristic times of the Auger decay pathways. Understanding these charge rearrangement processes and, in particular, the complex electronic and nuclear dynamics created by multiple ionization that lead to nuclear motion on the ultra-short time scale of the photon pulse are crucial for coherent x-ray diffraction imaging schemes based on the so-called diffract-before-destroy concept [20, 21]. In this context, the electronic and nuclear dynamics are often referred to as radiation damage since they destroy the initial structure of the system which shall be investigated. Experimental studies on polyatomic molecules containing one heavy atom constituent, which results in strongly localized photo-absorption, open an intuitive way to bridge the gap between our understanding of the single-atom response to intense, ultra-fast x-rays and the behaviour of larger nano-scale systems. They also shed light on mechanisms of radiation damage and their characteristic time scales, in particular, in the vicinity of high-Z impurities [22], which is important for the novel phasing methods in x-ray crystallography at XFELs [23].
Further motivation for this kind of study is provided by the vision of XFEL-based single-molecule imaging experiments that aim at probing evolving molecular structures with atomic spatial and femtosecond temporal resolution (e.g., via isolated-molecule x-ray diffraction [24], or via photoelectron diffraction and holography [25, 26]). Here, information on the molecular orientation for each single XFEL shot is needed in order to sort or correct single-shot spectra and to produce significant integrated signals that are not averaged over the typically random orientation of the molecules in the gas-phase. To date, many avenues are pursued to determine or control molecular orientation in the experiment, either by actively forcing the molecules into a certain orientation, e.g. by external fields such as a strong laser field created by femtosecond or nanosecond IR lasers [11, 24, 27–29], by determining the orientation of each molecule in the data analysis, e.g. by finding common lines in diffraction patterns [30] or by looking at ionic fragments of the molecule that are ejected with high kinetic energy. By inspecting the emission angles of these fragments in the laboratory frame, it can be possible to determine the orientation of the molecule a posteriori on a shot-to-shot basis. This technique, which is based on the coincident detection of fragment ions with other observables, has already been pursued for many years in photoionization studies at synchrotron sources [31–37], in femtosecond spectroscopy of photochemical reaction dynamics [38–40] and in collision physics experiments (see [41] and references therein).
In accordance with the above discussion, the main goal of the present paper is twofold. First, it extends our previously reported study on methylselenol (CH3SeH) molecules [19] to a larger selenium-containing system, ethylselenol (C2H5SeH), and investigates how the charge rearrangement mechanisms in the molecular environment depend on the size of the molecule. Second, it presents a detailed analysis of angular correlations in the emission of coincident ionic fragments and discusses to what extent the emission direction of these fragments can serve as an indication for the orientation of polyatomic molecules in space. We show that these coincidence techniques can also be applied at XFELs and present the first experimental results on the determination of the molecular geometry and molecular orientation after multiple inner-shell ionization by the XFEL by means of coincident three-dimensional (3D) ion momentum spectroscopy. By investigating kinetic energies of the fragments and angular correlations between multiple fragments measured in coincidence, the information on both bond lengths and orientation can be obtained and proton angular distributions can be investigated in the molecular frame defined by the heavier atom bond. In the case of methylselenol, this allows disentangling the otherwise undistinguishable protons from the selenium and carbon sites.
2. Experimental details
The experiment was performed at the atomic, molecular, and optical science (AMO) beam line [42] of the LCLS at the SLAC National Accelerator Laboratory using the CFEL ASG multi-purpose (CAMP) end station [43, 44]. For this experiment, the pnCCD x-ray photon detectors [43] were removed from the CAMP setup and a base pressure of ∼2×10−10 mbar was reached in the reaction chamber.
Figure 1 shows a schematic of the experimental setup with the continuous supersonic molecular beam of either methylselenol or ethylselenol molecules intersected by the XFEL beam inside a 3D-momentum-resolving ion spectrometer. The setup was identical to the one briefly described in our previous publication on methylselenol [19]. In the inset of the figure the geometries of both investigated molecules are sketched.
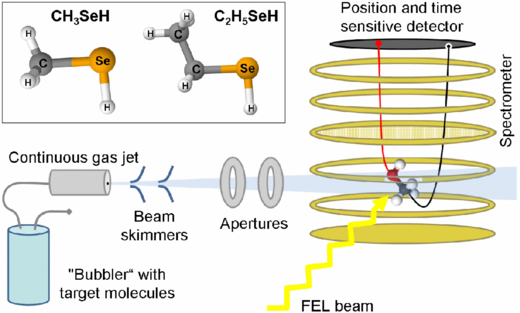
Figure 1. Overview of the spectrometer and the target injection setup in the CAMP chamber [43] used to perform coincident ion momentum spectroscopy on small organic molecules ionized with an x-ray FEL. The molecules are injected into the vacuum in a continuous supersonic molecular beam and are ionized by the focused XFEL beam inside a spectrometer that allows coincident 3D-momentum-resolved detection of fragment ions emitted in the full 4π solid angle on a delay line detector (see text for details). Inset: sketch of the geometries of methylselenol and ethylselenol molecules.
Download figure:
Standard image High-resolution imageIn order to prepare a cold, spatially well-defined and dilute target, the molecular beam produced by continuous supersonic expansion through a 30 µm aperture was doubly skimmed (diameter of first skimmer: 180 µm; diameter of second skimmer: 400 µm) and additionally collimated and separated from the main chamber by two differential pumping stages with 3 mm diameter round openings. Helium carrier gas (at 1 bar backing pressure) was sent though the 'bubbler' containing methylselenol and ethylselenol, both liquid at room temperature. After intersection with the XFEL pulses, the molecular beam was collected by a two-stage differentially pumped jet dump.
The XFEL pulses with photon energy of either 1.7 or 2 keV had a nominal pulse duration of ∼5 fs FWHM and were focused to a ∼10 µm2 spot. To achieve these ultra-short pulses, LCLS was operated in low bunch-charge mode [45] which yielded typical pulse energies around 0.4 mJ. Assuming a beamline transmission of 35% [6], the intensity at the interaction point reached ∼4×1017 W cm−2.
A constant electric field of 400 V cm−1 inside the ion spectrometer, which consisted of a 90 mm extraction region and a 180 mm field-free drift region, each separated by a high transmission grid to prevent field distortions, projected the ionic fragments produced by the interaction of the XFEL pulses with the molecules onto an 80 mm time- and position-sensitive microchannel-plate (MCP) detector equipped with a Roentdek delay-line anode. This field strength was necessary to ensure the full 4π solid acceptance angle of singly charged ions with energies of up to 100 eV. From the ion times of flights provided by the MCP and the hit positions provided by the delay line for each of the detected particle, the 3D momentum vectors of multiple coincident ionic fragments were reconstructed. Details of this ion momentum imaging technique under typical FEL conditions can be found in [46].
For small molecules, the final charge state of the molecular system after interaction with the XFEL pulse can, in principle, be identified by detecting all ionic fragments produced from the same molecule in coincidence, provided that experimental conditions are chosen such that no more than one molecule is typically ionized per XFEL pulse. However, given ion detection efficiencies of considerably less than 50% (defined by the MCP detection efficiency and combined grid transmission), such coincidences up to six-fold (for methylselenol) or nine-fold (for ethylselenol) are definitely out of reach, especially in view of the LCLS repetition rate of 120 Hz. Therefore, the data presented here were obtained by analyzing double coincidences (for methylselenol) or triple coincidences (for ethylselenol) between the heavy carbon and selenium ions. For methylselenol, triple coincidence data between carbon, selenium and one additional proton were also analyzed.
3. Results and discussion
3.1. General considerations
As several previous publications studying both single atoms [3, 5–8] and molecular systems [9–15, 19] have shown, the ionization process at the pulse intensities that can be reached at current XFEL facilities proceeds mostly by sequential inner-shell photoionizations and subsequent Auger decays, while direct ('non-sequential') two-photon absorption was found to be rather small [4, 47]. In methylselenol and ethylselenol molecules, the target systems chosen in this work, the photon energies of 1.7 and 2.0 keV mainly result in photoionization of the selenium L-shell with cross sections of 0.7 and 0.4 Mb, respectively. Since the cross sections for photoionization of the selenium M- and N-shells and of the carbon K-shell and valences are significantly smaller at these photon energies (≤0.01 Mb), this results in an initial photo-absorption almost fully localized at the selenium L-shell. For heavy atoms like Se or Kr a vacancy in the L-shell leads to several relaxation steps in the form of an Auger cascade. Kr, which has an electronic configuration resembling that of Se and a similar photoionization cross section, is therefore chosen in this work for comparison to the Se-containing molecules. The first relaxation most likely happens by intra-atomic LMM Auger (hole lifetime ∼500 as) on the Se site producing mainly 3d (and less likely 3p) vacancies. This is followed by the decay of these vacancies involving the molecular valence electrons (MNN Auger) which occurs on a ∼10 fs time-scale [48, 49]. The latter step causes a considerable portion of the total charge to be distributed over all molecular constituents [19] even though the initial inner-shell photo-absorption and the first Auger decay are very likely fully localized on the selenium atom.
As a result of the (multiple) inner-shell ionization at the Se L-edge and the subsequent Auger decays, the molecules become highly charged and consequently break up into multiple ionic fragments. For the simpler case of methylselenol molecules, this reaction schematically proceeds in the following way:
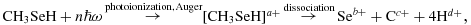
where the variables a, b, c, d are integer numbers fulfilling a = b + c + 4d. Note that b, c and d may include zero, meaning that also neutral fragments can be produced.
As shown previously for the cases of methylselenol [19] and nitrogen [13], the reaction does not proceed strictly sequentially and is governed by an intricate interplay between the time scales of the multiple photoionization events and Auger decay cascades and significant nuclear motion that already occurs on the same time scale. The following results and discussion are meant to provide further insight into the details of these complex reactions.
3.2. Ion time-of-flight spectra, ion–ion coincidence spectra and total charge state distributions
Figure 2(a) shows the ion time-of-flight spectra resulting from the ionization and fragmentation of methylselenol and ethylselenol molecules by LCLS pulses with a nominal pulse duration of 5 fs, a nominal pulse energy of 0.4 mJ, and a photon energy of 2.0 keV (methylselenol, red dashed line) and 1.7 keV (ethylselenol, blue solid line). Note that the actual pulse energy on target is lower since the beamline transmission on the order of 35% [6] is not included in the number given above.

Figure 2. (a) Ion time-of-flight spectra resulting from the ionization and fragmentation of methylselenol (CH3SeH) and ethylselenol (C2H5SeH) molecules by 5 fs-XFEL pulses with a nominal pulse energy of 0.4 mJ and a photon energy of 2.0 keV (methylselenol, red dashed line) and 1.7 keV (ethylselenol, blue solid line). The two spectra are normalized on the Se1+ peak such that differences in the other peak heights represent relative differences with respect to Se1+. The changes in the background signal (indicated by the arrows) are due to the higher residual gas background in the methylselenol spectrum. (b) Photoion–photoion coincidence (PIPICO) spectrum of methylselenol taken under the same experimental conditions as in (a). The two insets are zooms into low (green) and high charge channels (red). For strong channels, the five main isotopes of selenium can be identified in the spectrum as distinct parallel lines.
Download figure:
Standard image High-resolution imageThe two spectra are normalized on the Se1+ peak such that differences in the other peak heights represent relative differences with respect to Se1+. Here it should be noted that the shoulders appearing on the left side of Se1+ and on the right side of Se2+ peaks result from the residual gas ionization (most likely contaminations by larger hydrocarbons) and not from the target molecules. This was verified considering the position offset characteristic to all fragments originating from the supersonic gas jet. Comparing the spectra for the two different molecules, a significantly higher abundance of Se2+, Se3+, Se4+ and C2+ relative to Se1+ can be observed for methylselenol compared to ethylselenol, while the relative yield of C1+ is slightly higher in ethylselenol. Apart from the fact that the ethylselenol data were taken at a lower photon energy for purely technical reasons, which increases the total ionization probability since the Se L-shell photoionization cross section at 1.7 keV is higher than at 2.0 keV, the only difference between the two molecules is the addition of one carbon atom and two hydrogen atoms in the case of ethylselenol as compared to methylselenol (see the inset of figure 1). From the inspection of the ion time-of-flight spectra, we can therefore conclude that fewer multiply charged fragments are produced from ethylselenol since the total charge in the molecule can be distributed over a larger number of constituents.
The photoion–photoion coincidence (PIPICO) spectrum of methylselenol fragmentation is displayed in figure 2(b). The diagonal lines observed in the PIPICO spectrum reflect the momentum conservation between the carbon and selenium ions of different charge states. Since the momenta of the protons/hydrogen atoms are considerably smaller, they only result in a broadening of the lines (compared, e.g., to the case of diatomic molecule fragmentation [46]). For strong channels, contributions due to a few (up to five) main isotopes of selenium can be clearly identified in the spectrum as distinct parallel lines.
From this coincidence spectrum and the corresponding triple coincidence (PIPIPICO) spectrum for ethylselenol (not shown here), it is possible to determine the relative yields of the different selenium–carbon coincidence pairs or selenium–carbon–carbon coincidence triplets, respectively, which are presented in figures 3(a) and (b) and the total charge state distributions shown in figures 3(c) and (d). In the latter two, the sum of Se and C charges measured in coincidence is denoted as 'heavy ion charge', while the total charge of the molecule assuming that four or six H+ ions were produced is denoted as CH3SeH and C2H5SeH charge, respectively. While this assumption is well justified for the fragmentation of the most highly charged molecules [50], deviations are expected for the channels with lower heavy ion charge, especially for the ethylselenol molecule.
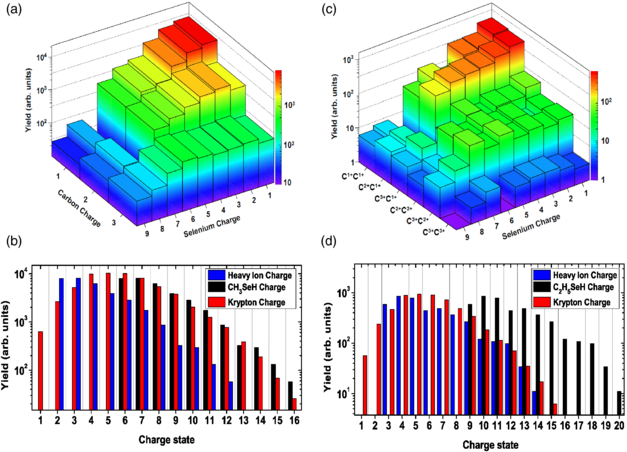
Figure 3. Top panels: measured yield of Se and C ion coincidences for (a) methylselenol at 2 keV and (c) ethylselenol at 1.7 keV photon energy. For ethylselenol, the yields of triple coincidences of Se-ions with both carbon ions (the combinations are indicated at the axis) are shown. Note that the Se7+C1+ and Se7+C2+ coincidence channels in methylselenol and all coincidence combinations of Se7+ with C1+ in ethylselenol had to be omitted in the analysis due to significant contributions of false coincidences from the residual background gas. Bottom panels: the total charge induced on the (b) methylselenol and (d) ethylselenol molecules compared to the charge state distribution observed in krypton at 2 keV photon energy under the same experimental conditions. The heavy ion charge represents the sum of Se and C charges measured in coincidence, whereas the CH3SeH and C2H5SeH charge denotes the total charge of the molecule assuming that four or six H+ ions were produced, respectively.
Download figure:
Standard image High-resolution imageNevertheless, clear trends can still be derived from the comparison of the molecular charge state distributions to the charge state distribution of isolated krypton atoms at 2.0 keV, which is also shown in figures 3(b) and (d). Kr has a similar electronic configuration and absorption cross section (0.5 Mb at 2.0 keV) to Se, and therefore serves as a model to estimate ionization without the molecular environment. As noted earlier [19], the maximum total charge induced on the methylselenol molecule and the overall charge state distribution for this molecule is very similar to the atomic case while the individual charge of the Se atom is much lower than for Kr, which is a clear indication of charge redistribution in the molecular environment.
In contrast to this, figure 3(d) for ethylselenol shows a significantly higher total charge than both methylselenol and krypton, even though the relative yields of the individual higher charged ions (Se2+, Se3+, Se4+) compared to Se1+ is reduced, as seen in the non-coincident ion time-of-flight spectrum discussed above. For ethylselenol, the 'heavy ion charge' distribution already shows a similar shape to the Kr charge state distribution, while the total charge of the molecule clearly exceeds the case of the isolated atom as well as that of methylselenol, even if one takes into account that, most likely, not all six hydrogen atoms are emitted as protons. Some of this effect can be attributed to the higher photoionization cross section for (neutral) selenium at 1.7 keV photon energy as opposed to methylselenol and Kr at 2.0 keV, but we suspect that an additional contribution comes from the fact that there are considerably more (valence) electrons in ethylselenol, which leads to more efficient Auger cascades that can reach more highly charged final states. Unfortunately, these two possible causes cannot be disentangled in the present study and their resolution will require further experimental and/or theoretical investigations.
3.3. Fragment energies
In addition to measuring ion time-of-flight and PIPI(PI)CO spectra, our 3D-momentum resolving spectrometer allows for precise determination of the fragment ion kinetic energies for all combinations of coincident fragments, as already shown in a previous publication [19]. In figures 4 and 5, we present a selection of kinetic energy distributions (KED) of singly and doubly charged carbon ions resulting from methylselenol (figures 4(a) and (b), and figure 5(a)) and ethylselenol (figures 4(c) and (d), and figure 5(b)) fragmentation for different final charge states (defined from the coincident measurements). In order to understand how these results relate to the geometry of an unperturbed molecule, we performed a simple simulation of molecular Coulomb explosion.

Figure 4. The kinetic energy distributions of carbon ions for different fragmentation channels of methylselenol (a), (b) and ethylselenol (c), (d). Vertical lines of the same colour indicate simulated Coulomb explosion energies for the given charge states assuming the equilibrium geometry of the respective neutral molecule.
Download figure:
Standard image High-resolution image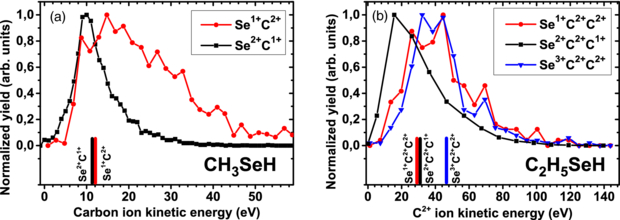
Figure 5. The kinetic energy distributions of carbon ions for 'anomalous' fragmentation channels where carbon is charged higher than selenium. For both, methylselenol (a) and ethylselenol (b), these channels are compared with the typical final states, where selenium is charged higher than carbon. Vertical lines of the same colour indicate simulated Coulomb explosion energies for the given charge states assuming the equilibrium geometry of the respective neutral molecule.
Download figure:
Standard image High-resolution imageTo simplify the simulation, we do not include the photoionization step nor any subsequent relaxations or other electronic effects. Instead, we assume all ionization processes to happen instantaneously and start the simulation with a set of ions with charges corresponding to the final (measured) charge states, and place them at the positions of the atoms in the equilibrium geometry of the neutral molecule.
The forces on the individual ions are determined by purely Coulombic interaction, and time propagation is given by a set of differential equations which are solved by a fifth-order Runge–Kutta method using Dormand–Prince parameters [51]. All particles are treated purely classically, and an adaptive time-step size ensures small numerical errors of positions and particle velocities while propagated towards their asymptotic values at the particle detector.
The simulation is thus designed to be fast and simple and to provide a rough idea of the ion kinetic energies (and ion emission directions) that are to be expected from an ultra-fast charge-up by intense FEL light-pulses followed by ultra-fast Auger decay. In particular, it does not include any geometric changes of the molecule during the multiple ionization process. Any deviations from the modelled kinetic energies and/or ion emission directions can therefore be interpreted as evidence for nuclear motion during the multiple ionization process.
Additional details of the model can be found in [52]. The results of the simulation are shown as vertical lines in figures 4 and 5.
In general, the KEDs for both molecules show a similar overall trend as discussed in [19] for methylselenol. As can be seen from figures 4(a) and (c) for the lowest charge states, the maximum of the KED fits well to the prediction of the Coulomb explosion model, indicating that there is no significant motion during the time needed to reach these final states. Since they are most likely produced by a single-photon absorption followed by one or two ultra-fast (sub-fs) Auger decay steps, this behaviour can be readily understood. However, considering higher charge states for both molecules, one can clearly see that the simulation starts to increasingly overestimate the measured kinetic energies, indicating that the final charges are reached at internuclear distances considerably larger than the equilibrium one. As we have shown for the case of methylselenol [19] considering intensity-dependent yields of different charge state combinations, the channels where more than two or three electrons are removed from Se and C fragments are typically produced by two- or multi-photon absorption. Thus, the deviations of the measured ion kinetic energies from the Coulomb explosion model for these higher charge states reflect the nuclear motion within the time defined by the delays in the sequential photo-absorption steps (inverse of photo-absorption rate) and Auger decay processes involving the valence electrons. Since the x-ray pulses used here are of the order of ∼5 fs long, these time scales are comparable for the charge states with a few electrons removed, whereas the Auger time scale starts to dominate for the higher charge states reaching tens of femtoseconds or more ([48], see also [7, 49] for Kr).
Although this behaviour is observed for both, methyl- and ethylselenol, the deviation from the Coulomb explosion model is more apparent for the smaller molecule, where the simulated kinetic energies for the highest charge state combinations even start to run out of the high energy flank of the KED (see figure 4(b)). In ethylselenol, this is less pronounced because of the contribution from the third heavy particle into the total momentum balance which causes the Coulomb explosion energy for a given total charge of the system to be distributed along two reaction coordinates instead of one (Se–C) for the case of methylselenol. The contribution of the third heavy fragment also explains the overall broader KEDs for ethylselenol fragments. As was shown in [19], the protons typically leave the system in the early ionization stage and do not influence the energies of heavier particles significantly.
In [19] we have also shown that for channels where the carbon fragment in the final state is charged higher than the selenium, very broad KEDs and extremely high fragment energies can be observed, which considerably exceed the predictions of the Coulomb explosion model for methylselenol. This is illustrated in figure 5(a), where the KEDs for Se2+ + C1+ and Se1+ + C2+ channels are compared. Although the simulation predicts similar explosion energies for both final states (note that according to the simulation, this is true independently of the proton charge), the measured distribution for the Se1+ + C2+ channel extends to much higher energies. In fact, most of the events have energies well beyond the Coulomb explosion limit. We attribute this behaviour to the creation of a transient highly charged Se–C complex and subsequent charge transfer to the hydrogen atoms. Although this hypothesis cannot be strictly proven by the present experimental data, it provides the most plausible explanation for the anomalously high fragment energies observed. Recent experiments on heavy diatomic molecules which do not contain hydrogen atoms further confirm this explanation [53].
Under the above assumption, the charge transfer to the hydrogen atoms needs to occur after the (initially highly charged) heavy fragments have acquired their final kinetic energies. On the other hand, the measured energies of the protons detected in coincidence with the 'anomalous' channels where qC > qSe are considerably lower than the simulated Coulomb explosion values [19, 51] suggesting that they have moved significantly before the final charge state of the system has been reached. Therefore, one of the central questions that remains open concerns the time and length scales of the observed charge redistribution, i.e. how much the hydrogen fragments moved by the time when the charge transfer occurs.
As can be seen from figure 5(b), a similar effect is observed for the ethylselenol molecule. Here, the C2+ fragment energy for the Se1+ + C2+ + C2+ final state considerably exceeds the one for the Se2+ + C2+ + C1+ final state, even though the total charge of the system is the same and the Coulomb explosion model yields almost identical values. As a matter of fact, the KED for the Se1+ + C2+ + C2+ channel is very similar to the one of the Se3+ + C2+ + C2+ state which has a considerably higher total charge. This again corroborates our hypothesis of a transient highly charged Se–C complex and subsequent charge transfer to the hydrogen atoms.
Such a delayed charge transfer from the Se–C complex to the hydrogen atoms is expected to occur, with certain probability, for essentially all channels and is most likely responsible for the rather large widths of the KED spectra, in particular, for the high-energy tails of the KEDs. However, the final state channels where the weakly absorbing carbon atoms are charged higher than the strongly absorbing selenium have, statistically, undergone the most charge redistribution within the molecule and the signature of the delayed charge transfer to the hydrogen atoms is thus most pronounced here.
3.4. Vector correlations and determination of molecular geometry and orientation
By measuring the 3D momentum vectors of multiple fragments in coincidence, we are also able to investigate vector correlations, i.e. correlations between the emission directions of the various fragments relative to each other. This yields information about the fragmentation mechanisms and fragmentation time scales (see e.g. [54–57]). It also provides data that could allow one to determine the orientation of certain molecular axes in space [31–37]. Here, we explore to what extent this technique, when applied to multiple inner-shell photoionization by intense XFEL pulses, is able to provide information on the molecular orientation on a shot-to-shot basis.
3.4.1. Methylselenol
Figure 6(a) shows the angular correlation between carbon and selenium ion momentum vectors, i.e. the measured distribution of angles between C and Se ions detected in coincidence. For all combinations of C and Se ion charge states, the distributions are peaked around cos(αSe,C) = −1 corresponding to an angle αSe,C of 180° between the momentum vectors of the C and Se ions. In other words, the two heavy ions are almost always emitted 'back-to-back', similar to the fragmentation of a diatomic molecule. This back-to-back emission is more pronounced the higher the C and Se ions are charged, while the distribution is rather broad for coincidences of Se1+ with C1+. Qualitatively, this behaviour can be easily rationalized since the contribution of the relatively light protons to the total momentum is rather small.

Figure 6. (a) Measured distribution of angles between C and Se ion momentum vectors detected in coincidence for methylselenol (CH3SeH). The distributions are peaked around cos(αSe,C) = –1 corresponding to 180° between the momentum vectors of the ions ('back-to-back' emission). α is the angle between the momentum vectors of the ionic fragments. (b) Angular distribution of protons relative to the Se–C axis as defined by the direction of the carbon ion momentum. The peaks centred around cos(αC,H) = –0.4 and cos(αC,H ) = + 0.4 correspond to the Se-H and the C-H angles of 114° and 66°, respectively. The integrals of the two peaks also show a 3:1 ratio corresponding to the numbers of protons bound to carbon and selenium. (c), (d) Kinetic energy distributions for protons with C and Se neighbours, respectively, detected in a triple coincidence with (c) Se2+ and C2+ or (d) Se5+ and C2+ .
Download figure:
Standard image High-resolution imageGiven the rather strict back-to-back emission of all but the lowest charge combinations of heavy fragments, the emission direction of these fragments is a good marker for the orientation of the Se–C axis at the time of fragmentation (since the pulse duration as well as the Auger lifetimes are short compared to the rotational period). In figure 6(b), the emission angle of the protons is thus plotted relative to the momentum vectors of the carbon ions stemming from Se2+–C2+ and Se5+–C2+ coincidences. The resulting distribution shows two clear maxima at cos(αC,H) = −0.4 and cos(αC,H) = + 0.4 (corresponding to angles of 114° and 66°) that can be attributed to the hydrogen atoms bound to the selenium and carbon (left and right peak), respectively. The integrals of the two peaks also show a 3:1 ratio corresponding to the numbers of hydrogen atoms bound to these species. This also supports the assumption made earlier that all hydrogen atoms are emitted as protons during the fragmentation. The width of the peaks reflects the width of the Se–C angle distribution plus any additional angular motion of the proton during the multiple ionization.
Compared to the bond angles of the hydrogen atoms in methylselenol in the equilibrium geometry given in the literature [58], which are 70.0° for the hydrogen atoms next to carbon and 95.5° for the hydrogen atom next to selenium, the measured angles for the carbon-site protons are in good agreement with the equilibrium geometry, whereas the measured angles for the protons from the selenium-site are larger than expected. This is due to the nature of the Coulomb explosion, where each charged particle interacts with all its charged neighbours. Because of the geometry of the molecule, with a close to 90° bond angle of the C–Se–H site and bond lengths d(H–Se) = 1.53 Å and d(C–Se) = 1.97 Å, the distance between the Se proton and the carbon is only 2.49 Å. This leads to a significant repulsion between the 'selenium' proton and the carbon charge, thus producing a larger angle between the emission direction of this 'selenium' proton and the carbon. A similar effect is also discussed in a study of UV-photo dissociation of H2S [59].
By selecting those protons whose momentum vectors have angles relative to the Se–C axis with negative or positive cosine value, we distinguish protons neighbouring the selenium or carbon site, respectively. The KED of these site-selected protons for two sets of Se–C charge states, Se2+ C2+, and Se5+ C2+, are plotted in figures 6(c) and (d). Surprisingly, no difference is observed in the high-energy part of the spectra, indicating that protons are emitted long before the final state charge is reached. In the low-energy part of the spectra, considerable enhancement of the low-energy feature (between 10 and 20 eV in figure 6(c) and centred at 20 eV in figure 6(d)) is found for protons emitted at the Se site. According to the simulation, this would correspond to a proton emitted from a singly or doubly charged Se and a neutral C atom, respectively [52]. This distinct feature, which can be observed for essentially all final charge state combinations, suggests that the low-energy part of the proton KED reflects the initial step in the sequential build-up of the total charge of the molecule, whereas the high-energy tail is due to the channels where the whole molecule is charged fast before significant nuclei displacement occurs.
3.4.2. Ethylselenol
Investigating the fragmentation of the larger ethylselenol molecule, we now have to consider a breakup into three heavy fragments, one selenium and two carbon ions. For such a nonlinear polyatomic molecule, the angular correlations of the fragment momentum vectors no longer reflect the molecular geometry directly but rather the sum of the Coulomb forces produced by all charges situated on the individual ions. In principle, these can be compared to predictions from a Coulomb explosion model, although the angular correlations, even more so than the fragment ion KEDs, are very sensitive to changes of the molecular geometry that occur during the multiple ionization process.
Maps of the angular correlations between the selenium and the two carbon ions for four representative channels (Se1+ + C1+ + C1+, Se2+ + C1+ + C1+, Se1+ + C2+ + C1+ and Se5+ + C2+ + C1+), which are obtained by measuring in coincidence the 3D momentum vectors of the three heavy ions that are created from an ethylselenol molecule, and are shown in figure 7 together with the respective simulated angles.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 7. Angular correlations between the three heavy-ion fragments of ethylselenol (C2H5SeH) showing the angle between selenium and singly charged carbon ion momentum vectors (x axis) versus the angle between the momentum vectors of the two carbon ions (y axis) in a triple coincidence. The respective angles produced by the Coulomb explosion simulation are shown as black dots. The two black dots correspond to two possible pathways that cannot be distinguished in the measurement. In the case of Se1+C1+C1+ (a) and Se2+C1+C1+ (b) the two dots originate from the ambiguity of the Se–C angle for two indistinguishable C1+ ions. For Se1+C2+C1+ (c) and Se5+C2+C1+ (d), the two dots represent the outcome of two different simulations where the C1+ either starts from the position close to the Se or from the one further away.
Download figure:
Standard image High-resolution image{kind=link}
The simulated angular correlations for a given final charge state from our Coulomb explosion model gives two values for each channel (indicted by dots in figure 7). These two values correspond to two scenarios of molecular dissociation that cannot be distinguished by our experimental method. For channels with symmetric carbon charges, e.g. Se2+ + C1+ + C1+, the dots correspond to the angles of the Se momentum vector with either of the two carbon ions. For asymmetric channels, e.g. Se5+ + C2+ + C1+, where the two carbons have different charge, they correspond to the angle between the Se and C1+ ion for the two cases where the C1+ starts from the position closer to the Se or from the position further away, respectively.
Overall, a significant change of the predicted angular correlations occurs for the different channels, while the change in experimental data is much less dramatic. The experimental correlation maps display broad features whose maxima only shift slightly as a function of the final charge distribution.
A possible explanation for this observation could be that the fragmentation process starts already at an early stage in the ionization sequence and although ionization continues towards the detected final charge state, the angular distribution of ions is governed mainly by the initial phase of the dissociation that occurs at relatively low total charge states. This hypothesis is consistent with our observation described in section 3.3 that the ion kinetic energies for high final charge states are also significantly lower than expected.
Qualitatively, we note that the angular distributions for the Se2+ + C1+ + C1+ and Se5+ + C2+ + C1+ channels show somewhat sharper peaks than the Se1+ + C1+ + C1+ and Se1+ + C2+ + C1+ channels, where the combined charge of the two carbon ions is higher than the selenium ion charge. As noted in section 3.3, these latter channels also show broad KEDs with energies far above the Coulomb limit that we attribute to delayed charge redistribution during the fragmentation process.
A more quantitative interpretation of the observed angular correlations will not be attempted here as we conclude that our simplified simulations that do not include motion of the nuclei during the multiple ionization process are not suited to adequately describe the angular correlations of complex fragmentation channels, especially considering the ultra-fast charge exchange processes that may be involved.
Considering the small difference between the simulated angles for the two cases of carbon charge distribution, for example in the Se5+ C2+ C1+ channel, and the given broad experimental distributions, an unambiguous assignment of the original positions of the two carbon ions is very challenging and, with the statistics of the present measurements, not feasible. Angular-resolved analysis of ion yields as well as studies of bending motion in the molecule upon multiple ionization is, thus, not further pursued in this work.
4. Conclusion
We have studied the inner-shell ionization and subsequent fragmentation of methylselenol (CH3SeH) and ethylselenol (C2H5SeH) molecules by intense ultra-short x-ray pulses by means of coincident ion momentum spectroscopy. In accordance with our previous study on methylselenol [19], we observed clear signatures of charge redistribution across the molecule. We found that in the larger ethylselenol molecules, the charges are distributed over more atomic constituents, thus producing lesser-charged fragments, even though the absolute charge induced on the molecule is higher than for methylselenol. This might be a hint towards the behaviour typical for larger, extended systems [16]. Recent results on L-shell ionization of iodine in different molecular environments obtained at the SACLA XFEL facility in Japan indicate that these redistribution processes become considerably more efficient for larger molecules like, e.g., iodo-uracil [60].
By analyzing fragment ion kinetic energies and angular correlations, we can identify fingerprints of the initial molecular geometry in the ion emission patterns and can, in particular, determine the laboratory-frame orientation of individual small molecules on a shot-by-shot basis. We do observe, however, that some of the bond lengths, also those between the heavy atomic constituents, change considerably already on the few-fs timescale given by the duration of the photon pulse and the typical Auger decay times. The first photoionization and subsequent Auger decays trigger an elongation of the bonds or the dissociation of the molecule and, as a result, the internuclear distance increases. As the charge state is further increased by slower Auger decays, and possibly, by the absorption of a second (or further) x-ray photon, the internuclear distance has already increased, and ionic fragments reach their final charge states at internuclear distances significantly higher than those in the equilibrium geometry of the molecule. As a consequence, the Coulomb repulsion force is reduced compared to the scenario where the same charge states reached at the equilibrium distance, resulting in final kinetic energies of the fragments that are lower than those expected for 'vertical' multiple ionization at the equilibrium geometry.
Although for most of the cases, the assumption of instantaneous multi-electron removal and Coulomb explosion at the equilibrium internuclear distance typically yields the upper limit for the measured kinetic energies of the fragments, this is not the case for channels where carbon atoms are charged higher than selenium. These channels, whose charge distribution itself represents an indication for charge rearrangement within the molecular systems, exhibit broad KEDs with their maxima well beyond the predictions of the Coulomb explosion model. Since the effects of shortening of the bonds prior to dissociation are unlikely to play a role for systems with more than two electrons removed, the most likely interpretation is that for these channels, the heavy atoms in the molecule are initially charged significantly higher than their final-state sum charge, and that the excess charge is transferred to the hydrogen atoms only after the heavy fragments have reached high kinetic energies.
Finally, this work demonstrated that coincident Coulomb explosion imaging can provide useful information about the spatial orientation of the molecule even for the case of sequential few-photon induced fragmentation. This has been proven by revealing three-particle angular correlation in the fragmentation of methylselenol and ethylselenol. For the larger ethylselenol molecule, maps of angular correlations of triple coincidences between the heavier ions where demonstrated, while for methylselenol, the protons originating from the selenium and carbon sites could be distinguished. The angular distribution of the protons is rather broad, whereas the selenium and carbon atoms are essentially emitted back to back, and exhibit angular correlations which become sharper with increasing charge states. Thus, the ion angular patterns of the heavier fragments can be exploited to determine the molecular orientation for further applications, e.g., photoelectron spectroscopy in the molecular frame.
At present XFELs such as the LCLS operating at 120 Hz repetition rate, the techniques of coincident ion momentum imaging employed in this work, as well as the related ion-electron coincidence spectroscopy, which enable measurements in the molecular frame, are extremely challenging, as can be seen from the limited statistical significance of the three-particle correlation maps presented here. However, this will change dramatically with the advent of new XFELs such as the European X-FEL in Hamburg or NGLS in Berkley, where multi-kHz or even MHz repetition rates will become available. This will open up completely new dimensions for the coincident techniques, enabling, in particular, photoelectron diffraction and holography experiments on molecules 'fixed in space' by coincident angle-resolved detection of emitted ionic fragments.
Acknowledgments
Parts of this research were carried out at the Linac Coherent Light Source (LCLS) at the SLAC National Accelerator Laboratory. LCLS is an Office of Science User Facility operated for the US Department of Energy Office of Science by Stanford University. Preparation measurements were performed at DORIS (DESY) at beamline BW3. We acknowledge the Max Planck Society for funding the development and operation of the CAMP instrument within the ASG at CFEL. AR acknowledges the support from Office of Basic Energy Sciences, US Department of Energy; DR acknowledges support from the Helmholtz Gemeinschaft through the Young Investigator Program. KU is grateful to Ministry of Education, Culture, Sports, Science and Technology of Japan for support of the X-ray Free-Electron Laser (XFEL) Utilization Research Project and the XFEL Priority Strategy Program. HS, LC and SD acknowledge support from the Carlsberg Foundation. JK and ST acknowledge support from the excellence cluster 'The Hamburg Centre for Ultrafast Imaging - Structure, Dynamics and Control of Matter at the Atomic Scale' of the Deutsche Forschungsgemeinschaft. We are grateful to C Schmidt for technical support, to J M Rost for valuable discussions, and to the SLAC staff for their support and hospitality during the beamtime.