
Abstract
Carbon nanotubes are fascinating nano-objects not only from a fundamental point of view but also with respect to their remarkable properties, holding great potential in new materials design. When combined with organic molecules, these properties can be enhanced or modulated in order to fulfil the demand in domains as diverse as molecular electronics, biomaterials or even construction engineering, to name a few. To adequately conceive these hybrid materials it is essential to fully appreciate the nature of molecule–carbon nanotube interactions. In this review, we will discuss some relevant fundamental and applied research done on encapsulated molecules in carbon nanotubes. We will particularly focus on the weak and van der Waals interactions which rule the molecule–tube coupling. Therefore a small state of the art on the theoretical methods used to describe these interactions is presented here. Then, we will discuss various applications of molecular encapsulation, where we will consider structural, magnetic, charge transfer and transport, and optical properties.
Export citation and abstract BibTeX RIS
1. Introduction
The discovery of carbon nanotubes (CNTs) [1] has given rise to a wide range of fundamental studies and led to significant technological applications based on these new materials and their derivatives. Due to their exceptional structural and electronic properties, they constitute elementary bricks for designing the future nano-objects of our daily life [2]. They are mechanically very resistant and chemically stable, which make them ideal candidates to create new kinds of fibres, ultra-light materials for construction or even colloidal solutions for new paints. Electronically, CNTs can be considered as an extended π-electron system, allowing reactions with other π-electron systems like aromatic molecules through π-stacking interaction [3]. Therefore, their interesting electronic properties can be tuned or modified by their interaction with atoms [4] or molecules [5]. For example, atomic substitution in a CNT with different elements leads to gap opening or chemical doping with a strong effect on the electronic conductance [6]. As in the case of graphene, it is also interesting to adsorb organic molecules on the outer wall of a nanotube in order to functionalize it and obtain interesting properties for applications in molecular electronics for example [7–9].
In addition to this, nanotubes have the unique ability to encapsulate molecules. In this way, the interaction of the molecule with the inner wall of the nanotube will affect its electronic properties, such as transport or optical properties, leading to a great deal of potential applications in the electronics components of tomorrow. For example, it has been demonstrated recently that molecular magnets can be inserted into CNTs, forming a new approach to the construction of ultrahigh-density magnetic data storage devices [10, 11]. For catalysis purposes, the confined space in the nanotube can also give rise to chemical reactions between different compounds, thus isolating them from the rest of the solution [12–15]. In biology or medicine one can also find interesting properties such as drugs delivery to specific organs using the nanotube as a transporter [16, 17]. And yet we are only at the beginning of the exploration of their fantastic properties.
In this review, we will mainly focus on the theoretical aspects of molecular encapsulation in CNTs and present some experimental results. An important difference between molecular encapsulation with respect to molecular adsorption or atomic substitution in a CNT is that this process is ruled by weak and van der Waals (vdW) interactions [18]. By weak interactions, we mean non-covalent interactions, like π stacking or physisorption. In any case, the encapsulation is a reversible process which does not alter the atomic structure of the nanotube. The goal lies in slightly modifying the nanotube properties through molecular insertion. From a theoretical point of view, which is the scope of this review, these weak and vdW interactions are really complicated to handle. Standard first-principles methods such as density-functional theory (DFT) [19, 20] are often used to model these kinds of systems, but the DFT formalism does not allow a correct treatment of vdW interactions. In some cases, error compensations yield to a reasonable result, but we will demonstrate here that it is not always the case and that the inclusion of such interactions is a requisite for an accurate characterization of these systems.
The structure of this review is the following: in the next section, I will present a state of the art of the theoretical methods which have been used to describe vdW interactions mainly for applications in encapsulated molecules in CNTs. Then, in section 3, I will present several studies dedicated to the determination of the structural properties of these systems. In sections 4, 5, 6 and 7 I will discuss relevant examples of encapsulated molecules in CNTs presenting interesting properties in the fields of magnetism, electronics, optics and biomaterials, respectively. We will then conclude this review and open some perspectives for future studies of these systems.
2. State of the art on vdW interaction modelling
2.1. General
In this section we will review the main methods which have been developed recently to treat the question of weak and vdW interaction at the microscopic level. We therefore present here the essential approaches related to DFT calculations. Other approaches, such as quantum chemistry methods, are very important for the description of vdW interactions. However, these methods are rather expensive with respect to the computational cost and, consequently, they have not been applied as often to describe molecular encapsulation in CNTs. For more general approaches, the reader is referred to review articles from Grimme or Dobson for example [21–23]. As is well known, vdW energy is associated to charge fluctuations in the material due to the coupling with the vacuum electromagnetic field, giving rise to non-permanent dipoles. Those non-permanent dipoles interact together yielding an attractive interaction energy, namely the vdW energy. Consequently, this is a field interaction not directly related with an overlap of the spatial electronic densities. This constitutes the main problem for the DFT treatment based on spatial electronic density approximations. For example, local density approximation (LDA) or generalized gradient approximation (GGA) fail totally to describe such interactions [19, 20]. Another way of considering the problem lies in the exponential decay of the DFT calculated interactions, due to the exponential behaviour of the wave functions overlaps, while in the mean time, the vdW interaction is rather long range. As a consequence, one can notice that it is really difficult to find a functional able to handle in the mean time short-range and covalent interactions on the one hand, and long-range and weak interactions on the other.
Consequently, in recent years, many approaches have been developed to overcome this difficulty in the frame of DFT, either as an intrinsic contribution or as an added external potential. Here we will present the main kinds of approximations, ranging from semi-empirical model, to full RPA implementation in DFT, through intermolecular perturbation theory. We will then discuss some applications of these different approaches, especially in the case of encapsulated molecules in CNTs.
2.2. From semi-empirical models to beyond DFT approaches
Many works have been devoted to the modelling of systems ruled by weak and vdW interactions, using semi-empirical approaches. The very first popular model was the Lennard-Jones potential:

where the parameters ε and σ are chosen empirically to fit the considered material. The r−6 term represents the vdW interaction as traditionally obtained from dipole–dipole interactions. The r−12 term is more arbitrary and is chosen to describe the repulsion at short range, whose origin is mainly the Pauli exclusion principle. This potential describes the interaction energy between two atoms at a distance r. To obtain the total binding energy of the system, one has to sum all the pair energies of the two subsystems, taking into account the geometry of the system through the interatomic distance. This empirical method has been successful in providing a unified and consistent description of properties which depend on weak chemical interactions and vdW dipolar interactions. But even though this potential is widely used and has given numerous interesting results, it presents a strong disadvantage: the parameters have to be adjusted for each considered material, and it cannot take into account structural modifications of this material at the atomic level. For example, the deformation of a molecule during an adsorption process, which obviously will affect the interatomic potential, cannot be described correctly through this potential. Moreover, the arbitrarity of the r−12 term often has to be compensated by the dipolar term, leading to a bad estimation of the r−6 term, the pure vdW energy [24, 25]. This feature can be seen in the table represented in figure 1, where the coefficient A and B, which we usually call C6 and C12, are related to equation (1) through C6 = 4εσ6 and C12 = 4εσ12.

Figure 1. Table presenting the various C6 coefficients (as defined in the text) used for vdW calculations in graphene–graphene, C60–C60 and C60–graphene. Reprinted with permission from [24]. Copyright 2000 American Physical Society.
Download figure:
Standard image High-resolution imageIt clearly shows that according to the considered system, the coefficients can be different for around 30%. However, this formalism has provided good results in the description of encapsulated molecules in nanotubes, as for example, C60 phases in various CNTs (figure 2).

Figure 2. Representation of different phases of C60 molecules encapsulated in CNTs. Reprinted with permission from [26]. Copyright 2003 American Physical Society.
Download figure:
Standard image High-resolution imageThis formalism was strongly improved in the work of Grimme et al [27–29] by substituting the C12 coefficients by a damping function, which is much more efficient to describe the repulsive part of the interaction and in overall to cancel the vdW contribution at short distance. Namely, the DFT-D formalism [27] is based on a standard DFT calculation where a dispersion term of the form
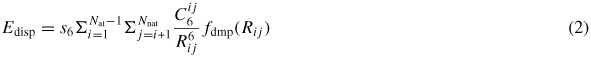
is added to the total energy of the system. Here, Nat is the number of atoms in the system,
 denotes the dispersion coefficient for atom pair (i, j), s6 is a global scaling factor and Rij is the interatomic distance. The damping function is used to avoid singularity at small distances and is given by
denotes the dispersion coefficient for atom pair (i, j), s6 is a global scaling factor and Rij is the interatomic distance. The damping function is used to avoid singularity at small distances and is given by

where R0 is the sum of atomic vdW radii.
Even though there are no results yet on molecular encapsulation in CNTs, this method is really useful and is still being used extensively for molecular systems. For example, we have also used this damping function in combination with our vdW calculation to determine the benzene–gold equilibrium distance accurately [30].
A following improvement to these approaches is the method developed by Tkatchenko et al [31, 32]. In this formalism, the vdW energy is written as
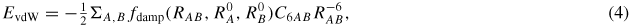
where RAB is the distance between atoms A and B, C6AB is the corresponding C6 coefficient,
 and
and
 are the vdW radii. The
are the vdW radii. The
 singularity at small distances is eliminated by the short-ranged damping function
singularity at small distances is eliminated by the short-ranged damping function
 . This approach also considers a Fermi-type damping function similar to the one proposed by Grimme et al. This approach is pretty similar to the one of Grimme et al, except that the C6 coefficients are no longer adjusted but determined from referenced polarizabilities and ground state electronic densities obtained with DFT calculations. This takes into account the atomic environment. This method represents an important step in the non-empirical determination of C6 coefficients for vdW forces.
. This approach also considers a Fermi-type damping function similar to the one proposed by Grimme et al. This approach is pretty similar to the one of Grimme et al, except that the C6 coefficients are no longer adjusted but determined from referenced polarizabilities and ground state electronic densities obtained with DFT calculations. This takes into account the atomic environment. This method represents an important step in the non-empirical determination of C6 coefficients for vdW forces.
The next step in the development from semi-empirical to first-principles methods to characterize accurately the weak and vdW interactions is the vdW-DF approach proposed by Langreth, Lundqvist and collaborators [33–36]. In this model, the vdW energy is described as
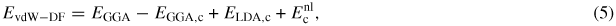
where the LDA correlation energy ELDA,c is substituted by the GGA correlation energy, EGGA,c, and where the non-local correlation energy
 is added. This non-local correlation energy is expressed as
is added. This non-local correlation energy is expressed as

where φ(r, r') is a function depending on |r − r'|, the charge density n and its gradient at the positions r and r'|, respectively. This formulation of the vdW energy is not yet a functional in the present form, but it has been implemented self-consistently as a functional in the SIESTA code [37]. This self-consistent version of the Lundqvist and Langreth functional has been applied to the structural determination of a double-wall CNT. The non-self-consistent vdW-DF has been applied in general for sparse matter characterization, including molecules and CNTs [38] as illustrated in figure 3.
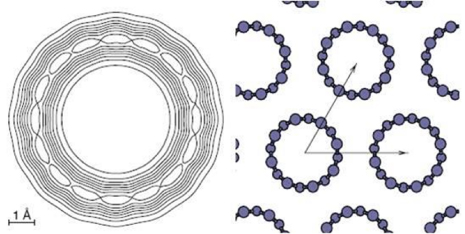
Figure 3. Illustration of a semiconducting CNT crystal for vdW-DF calculation. Reprinted with permission from [39]. Copyright 2008 American Physical Society.
Download figure:
Standard image High-resolution imageNotice that this approach has been used by several groups for the study of aromatic molecule adsorption on noble metal surfaces for example, in combination with semi-empirical corrections to accurately determine the molecule–metal equilibrium distance [40].
The last further step in the treatment of weak and vdW interactions in DFT is the development of methodologies based on the adiabatic-connection fluctuation-dissipation (ACFDT) [41–43] used directly in the random phase approximation (RPA). In this frame, the correlation energy (including vdW interaction) is written as
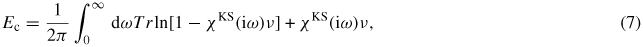
with χKS being the independent particle (non-interacting) Kohn–Sham (KS) response function, and ν being the Coulomb kernel. The exchange energy is calculated exactly using the Hartree–Fock expression. Calculations on graphene have been performed within this formalism, using the VASP code [44, 45].
Besides these developments, we can also cite the work of Silvestrelli et al [46] where the vdW energy is determined through the use of Wannier functions. Due to high computational costs, those last methods are rather prohibitive for the study of big systems like encapsulated molecules in nanotubes.
2.3. DFT and intermolecular perturbation theory: the LCAO-S2 + vdW model
In this part we will now present our treatment of weak and vdW interactions, i.e. an intermolecular perturbation theory approach within the frame of DFT. Namely, the LCAO-S2 + vdW model has been first developed for the study of rare gas dimers [47] and then to describe the interaction between two graphene planes [48, 49] and graphitic materials in general [50]. This approach is based on the LCAO-OO formalism. Details about this formalism can be found in [51–53].
As a starting point, we first study each isolated subsystem (for example a molecule and a nanotube, before encapsulation), in order to properly describe subsequently their interaction. The DFT computational scheme, as well as the theoretical foundations underlying our calculations—a very efficient DFT localized orbital molecular dynamics technique (FIREBALL)—have been described in full detail elsewhere [54–57], and here we will only summarize the main points.
We first analyse all the systems involved in this study by using a self-consistent version of the Harris–Foulkes LDA functional [58, 59], instead of the traditional KS functional based on the electronic density, where the KS potential is calculated by approximating the total charge by a superposition of spherical charges around each atom. The FIREBALL simulation package uses a localized optimized minimal basis set [60], and the self-consistency is achieved over the occupation numbers through the Harris functional [61]. Besides, the LDA exchange-correlation energy is calculated using the efficient multi-centre weighted exchange-correlation density approximation (McWEDA) [55, 56]. To these DFT calculations we add 'weak interactions', which can be seen as two opposite contributions. The first one, named 'weak chemical' interaction, is due to the small overlaps between electronic densities of the interacting subsystems. Therefore, this energy can be determined as an expansion of the wave functions and operators with respect to these overlaps. This expansion is based on a development in S2 (since in the weak-interacting case the overlaps are really small) of the S−1/2 term appearing in the Löwdin orthogonalization, which induces a shift of the occupied eigenenergies of each independent subsystem, and leads to a repulsion energy between them. This shift can be defined as follows:
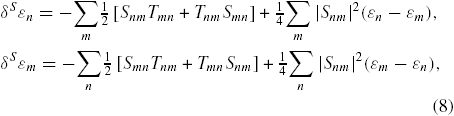
where Snm and Tmn are the overlapping and hopping integrals between eigenvectors m and n in the two interacting subsystems, for example the molecule and the nanotube. The effect of the hopping matrix elements, Tmn, can be calculated in a standard intermolecular second-order perturbation theory as
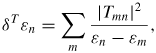

Thus, we obtain the following 'weak chemical' contribution to the interaction energy:
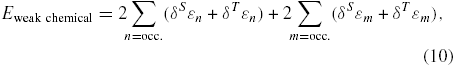
where a multiplicative factor of 2 has been included to take into account the spin degeneracy and only filled states are considered. Typically, in graphitic or hydrocarbonated materials this contribution is repulsive.
The second contribution is the pure vdW interaction, which finds its origin in charge fluctuations in each subsystems, arising from oscillating dipoles whose interaction gives the attractive part of the cohesive energy. This interaction is treated in the dipolar approximation, and added in perturbation to the total energy of the system. In this frame, the vdW energy can be written as
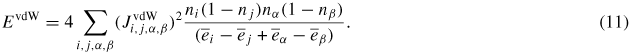
In this expression, ni are the orbital occupation numbers (per spin)

and
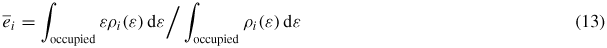
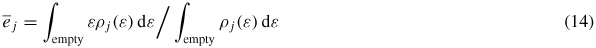
are average occupied and empty levels. The integral
 takes into account all the dipole–dipole excitations of the system. It is actually the result of the calculation of a four-centre Coulombic integral in the dipolar approximation and its corresponding expression is
takes into account all the dipole–dipole excitations of the system. It is actually the result of the calculation of a four-centre Coulombic integral in the dipolar approximation and its corresponding expression is

R is the distance between the two atoms assumed in this expression along the z-axis, which depends on the different dipolar matrix elements in each atom. Of course, this approximation would not be valid anymore if we would like to evaluate vdW interaction at really short distances, but this is not the case for this work. Moreover, as it was discussed above, this
 represents the bare vdW interaction, without screening, as approximated in our model.
represents the bare vdW interaction, without screening, as approximated in our model.
Using these expressions and the DFT band structures of each isolated subsystem obtained previously with FIREBALL, we can evaluate the intermolecular vdW energy. By combining this energy with the weak chemical repulsion obtained in the LCAO-S2 approach, we can determine the binding energy of the full system and compare it accurately with experimental results and other theoretical determinations. The balance of the two contributions gives the equilibrium configuration of the system. As an illustration, energetic calculations using this formalism for different structures of graphite are presented in figure 4 [49]. This formalism has been used in the example of molecular encapsulation in nanotubes that we will present in the next section. We have also used some extensions of this formalism to study the case of metal organics interfaces [30, 62].

Figure 4. Figure reprinted from [49] presenting (a) the stacking-fault energy surface (also called corrugation energy surface) of two interacting graphene planes. The square and circle symbols indicate the stationary points corresponding to the following high-symmetric structures. (b) The hexagonal, orthorhombic, rhombohedral and AA hexagonal stackings viewed perpendicular (above), parallel (below) to the c-axis. The energies of these structures with respect to hex-g are 1.66 meV/atom for ortho-g, 0.10 meV/atom for the rhom-g and 9.29 meV/atom for AA hex-g.
Download figure:
Standard image High-resolution imageAs a last remark, it is worth noticing that a general trend nowadays in the development of formalisms which take into account the vdW energy is the screening of the interaction. Indeed, this dynamical effect is mostly neglected even though it can actually reduce the C6 coefficients by up to a factor of 4. Going into detail on this topic is far beyond the scope of this review and moreover there is no relation yet with molecular encapsulation, but some approaches have been developed in [31, 63].
3. Structural properties of encapsulated molecules
In this section we now present some features of the structural aspects of encapsulated molecules in a CNT. As many hybrid molecule–nanotube systems have been explored, we only present here few representative examples which do not constitute an exhaustive overview of the domain. We will focus first on the peapod structures which have been widely studied, either experimentally or theoretically. In particular, we will present standard DFT studies without inclusion of weak and vdW interactions, and stress the differences when these interactions are considered. We will show an example in particular where demonstration is made that the inclusion of these interactions is a requisite for a correct description of the system, even though in some cases DFT results can give the good tendency by error compensations. Then we will show the structural properties of other molecules in nanotubes.
3.1. Encapsulation of C60 in a CNT: the peapod structure
Since the discovery of nanotubes and their ability to encapsulate molecules in their inner space, many studies have been devoted to these hybrid systems. The most famous and common example is the peapod structure [64], where C60 molecules are inserted in the nanotube, as illustrated in figure 5. This system is particularly interesting for the realization of true one-dimensional systems, were fullerenes are aligned inside the nanotube along one specific direction, due to the confinement inside the limited space of the nanotube. Consequently, the interaction between the fullerenes can be affected by the interaction with the nanotube. On another hand, the encapsulation of molecules inside the nanotube can lead to electronic modifications of the nanotube, as well as changes in its mechanical properties. Therefore, this hybrid system is of high interest for scientific and industrial applications.
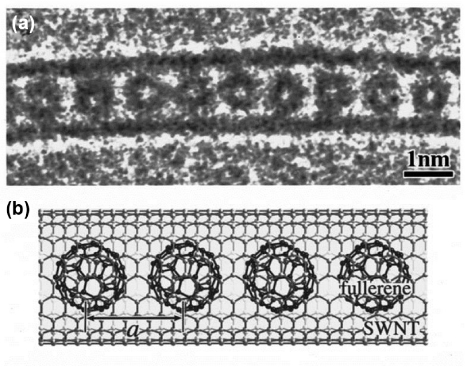
Figure 5. Representation of the so-called peapod structure. (a) High-resolution transmission electronic microscopy image and (b) its schematic illustration of an individual fullerene-encapsulated single-walled CNT [(C60)n@SWCNT]. Reprinted with permission from [65]. Copyright 2001 American Physical Society.
Download figure:
Standard image High-resolution imageSeveral models have been considered to characterize these systems. As an illustration, we can mention the recent work of Tang and Yang [66] who described the encapsulation of C60 molecules in SWCNT in solvent conditions by the mean of molecular dynamics simulations. In this approach, the vdW energy is included in the potentials used for the simulation and which have been fitted on reference materials earlier. Considering three different solvents, they have demonstrated the ability of inserting these molecules in the nanotube in good agreement with experimental results. In particular, they have shown that the insertion is not always possible due to a strong potential barrier at the entrance of the tube, stressing the important role of the solvent in molecular encapsulation in nanotube.
Coming back to first-principles methods, we have to say that many studies of encapsulated molecules in CNTs, and in particular peapods, are performed using standard DFT calculations without including weak and vdW forces. Although it seems to be a strong approximation, it can be justified in two ways: first, the emergence of methods able to treat this kind of interactions is very recent and, until they appeared, DFT calculations were considered as reference works even for weakly interacting systems. Second, in many cases, due to a compensation of errors, standard DFT can recover more or less the geometry of the system (with however a strong difference on the energy value), leading to a reasonable theoretical characterization of the peapod structure. This is the case for example of the work of Okada [67] where the energetics of C60 molecule inserted in CNTs ranging from the smallest diameter, (10, 10) to the largest, (20, 20), has been determined using DFT. It has been revealed that there is an important deformation of the nanotube with the inclusion of the fullerenes, which modifies the tube cross section from circular to elliptical shape. The corresponding deformation energy is around 10 meV per atom. In addition, the author has considered the case of the C60 dimerization in the nanotube, and found that this reaction is exothermic, the gain in energy being around 0.1 eV per C60. However, the activation barrier for the dimerization is found to be 1.4 eV indicating that polymerization is unlikely to take place without an external added energy like irradiation. Consequently, this work is really interesting for gaining an insight in the understanding of fullerene polymerization inside a CNT. However, when we have a look at the binding energy, we can notice that the lack of vdW energy changes the quantitative aspects of this system dramatically. For example, in this work, Okada finds a binding energy of around 1 eV per C60 molecule in a (12,12) nanotube.
Using the LCAO-S2+vdW approach, we have determined the binding energy of the encapsulated C60 in a (12,12) CNT [50]. The inclusion of vdW energy yields a binding energy of 2.28 eV per molecule, as represented in figure 6, which is more than twice of what is obtained in standard DFT calculation.
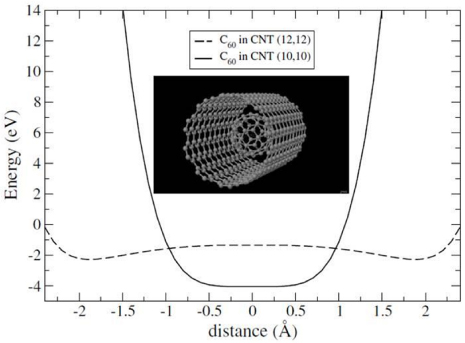
Figure 6. Binding energy of a C60 molecule encapsulated in a (10,10) and a (12,12) SWCNT as a function of the distance between the centre of the molecule and the SWCNT axis as calculated in [50]. Atomic configuration of the C60 in a (10,10) SWCNT is represented in inset.
Download figure:
Standard image High-resolution imageThis particular example stresses quite well the fact that even if DFT calculations can yield the correct geometry for those weak-interacting systems, the energetics are really dominated by vdW interactions. Therefore, their inclusion in the formalism is a requisite to obtain an accurate energetic description of the system.
In the next example, we will see that, in addition, there are weak-interacting systems where not only the energetics but also the geometry of the system are ruled by vdW interactions. In those cases, not including vdW energy in the formalism will lead to an erroneous result. The system is the following: we consider a C60 molecule encapsulated in a double-walled CNT (DWCNT) [68]. The inner tube is a (15,0) CNT meanwhile the outer tube is a (24,0), as represented in figure 7.

Figure 7. Atomistic model of a C60 molecule inside the DWCNT (15,0)@(24,0) used for the calculations. Figure reprinted from [68].
Download figure:
Standard image High-resolution imageThis system has been chosen in agreement with the experiments reported by Khlobystov et al [69] where assembled molecular arrays of C60 inside DWCNTs have been observed by means of high-resolution transmission electronic microscopy (HRTEM). The specificity of this system lies in the really small radius of the inner tube. Indeed, DFT calculations with or without inclusion of vdW forces show that C60 molecular encapsulation in the SWCNT (15,0) is not allowed thermodynamically due to the strong repulsion with the tube walls. The idea is then to stabilize the system using a second nanotube, the (24,0) around the C60@(15,0).
First of all, we can observe that the standard DFT-LDA calculation, performed with the FIREBALL code, yields a totally repulsive interaction, around 8.7 eV for the whole C60 molecule at the minimum of interaction, that is to say, in the centre of the DWCNT [68]. This result means that either the C60 molecule in the DWCNT(15,0)@(24,0) is not stable or the DFT-LDA formalism is unable to describe it properly since weak and vdW interactions are not considered. Now, we consider the evolution of the C60 molecule binding energy, calculated with the LCAO-S2 + vdW formalism, along the DWCNT axis. The purpose here is to observe the energy variation when moving the molecule inside and outside the tube. This binding energy is shown in figure 8.

Figure 8. C60 molecule cohesive energy in the (15,0)@(24,0) DWCNT using the LCAO-S2 + vdW formalism. The energy evolution is represented as a function of the molecule displacement along the DWCNT axis. Between 0 and 3.5 Å, the molecule is in the DWCNT and from 3.5 Å, the C60 molecule goes outside the DWCNT. The vertical red line indicates the end of the DWCNT. Figure reprinted from [68].
Download figure:
Standard image High-resolution imageAs we can observe, between 0 and 3.5 Å, there are oscillations of the binding energy, roughly between −1 and −0.5 eV/molecule. In this spatial range, the C60 molecule is located inside the DWCNT, and the oscillations correspond to the tube corrugation associated with the loss of interacting atoms when the molecule is getting closer to the tube entrance. At 0 Å, the binding energy value is 668 meV for the whole C60. Between 3.5 and 5 Å, we can see a potential barrier that the C60 molecule has to overcome to go out of the DWCNT. From 5 Å, we observe an increase of the binding energy which is explained by the weak interaction between the C60 molecule and the DWCNT entrance. This interaction presents an important energy minimum of almost 4 eV around 8.3 Å. This energy is much more important than in the DWCNT, because in this configuration the C60 feels much less repulsion from the tube than in the confined configuration. Consequently, the attractive part is not so well balanced, and the binding energy becomes bigger. The barrier energy to be overcome by the C60 molecule to go in the DWCNT is 3.426 eV in good agreement with previous work from Girifalco et al [24]. Farther from 8.3 Å, the binding energy begins to decrease, until there is no more interaction at very large distance. Experimentally, one of the proposed mechanisms entails that fullerenes enter nanotubes through defect openings which would then require less energy cost. Another mechanism would be (especially for DWCNTs) that fullerenes enter the tubes from their open ends after the DWCNTs are formed and for this a certain temperature is required. However, as far as we know the mechanism is not fully understood.
Consequently, in agreement with experiments the C60 molecule accommodation is confirmed when and only when the vdW interactions are included. This is an indication that results based only on standard DFT methods (whether LDA or GGA) can be misleading to predict the energetics for these systems if vdW interactions are not properly included. Finally this result stresses the fundamental importance of these kind of interactions, not only as DFT corrections, but as fundamental for the correct description of the physics of the system.
To close this part, we mention here an experimental work as an example of peapod extensions, which is the study of metallofullerene encapsulations in CNTs [70]. The authors have achieved the encapsulation of metallofullerene like La2@C80 and ErxSc3−xN@C80 (x = 0–3) and studied their structural properties by the mean of HRTEM and electron diffraction. Moreover, the thermal stability of these supramolecular assemblies has been characterized, which opens the way for the study of new electronic effects in such hybrid supramolecular structures. Similarly, on the theoretical side, we can mention the work of Ganji et al [71] who have considered the encapsulation of azafullerenes (nitrogen-doped fullerenes) on the basis of a DFT treatment. The obtained results are comparable to the one of standard peapods.
3.2. Encapsulation of flat molecules
In this part, we will now consider examples of more general molecules encapsulated in CNTs. In order to remain in the all-carbon systems, i.e. graphitic materials, we can consider the inclusion of graphene nanoribbons in SWCNT (GNR@SWCNT) [72]. In this study, Mandal et al characterize theoretically the encapsulation of coronene and perylene based nanoribbons in zig-zag SWCNT of different sizes, ranging from the (14,0) to the (20,0). The computational methodology is a self-consistent charge density-functional tight-binding (SCC-DFTB) method, considering vdW interaction between nanoribbon and nanotube (on the basis of a set of parameters for H and C), based on the Slater–Kirkwood model. Graphene nanoribbons have been placed in the centre of the tubes in order to perform geometry optimization and energetics calculations. As a result, the nanoribbon encapsulation in a nanotube is possible, as an exothermic process, which opens many experimental potentialities. However, if most of the systems remain with an unchanged shape, the smaller tubes have to get adapted to the size of the encapsulated nanoribbon. This aspect is obviously related to the repulsive energy between the tube wall and the nanoribbon when the tube is too small, leading to an unfavourable energetic situation. The most stable coronene based structure, GNR(Coronene)@(18,0), presents a metallic character while the other coronene based structures are semiconducting. Regarding the perylene based nanoribbons, the hybrid encapsulated systems appear to be of great interest for solar cell applications, due to a lower charge recombination rate.
Another prototypical system of graphitic-like molecules as a π-conjugated organic system is the molecule of benzene [73]. Its encapsulation in a zig-zag CNT (n, 0), with n ranging from 10 to 18, has been studied theoretically using the vdW-DF approach [35]. In this work, the position and orientation of the molecule inside the nanotube have been explored, in order to find the optimal tube diameter for encapsulation. This diameter appears to be around 1 nm. The wall–molecule distance depends on this diameter, but in all the cases, the molecule tends to align its molecular plane with the SWCNT axis. The chirality of the tubes is shown to play no role, but the tube has an influence on the intermolecular distances for confined benzene arrays. Here as well the structure and energetics of the hybrid system is ruled by vdW interactions.
In the same manner, we can mention the nice experimental work of Fujihara et al [15] where they report novel GNR growth through the vapour-phase doping of coronene in CNTs. The process is schematically represented in figure 9. The system is then characterized by HRTEM and Raman spectroscopy, revealing the encapsulation of linearly condensed coronene oligomers, that is to say GNRs. Consequently, this work shows that a CNT can constitute the host of molecular polymerization of graphitic molecules in order to grow GNR.
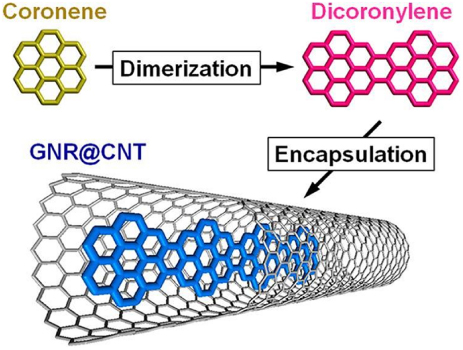
Figure 9. Graphical representation of the polymerization process from coronene inside a CNT to obtain a GNR. Reprinted with permission from [15]. Copyright 2012 American Chemical Society.
Download figure:
Standard image High-resolution imageSimilarly, in a theoretical study from Kim et al, the authors determine the energetics of a single acetylene (C2H2) molecule and the formation energy of acetylene polymerization inside the nanotube [74]. Using the PWSCF-DFT package, they calculate the activation energy barrier in polymerization of the acetylene molecules confined in a (5,5) nanotube, as represented in figure 10. The process is found to be exothermic since there is practically no activation energy barrier. As an application, a process is proposed to fabricate acetylene polymers in a CNT, without interchain coupling, helping to understand the intrinsic properties of the individual chains.
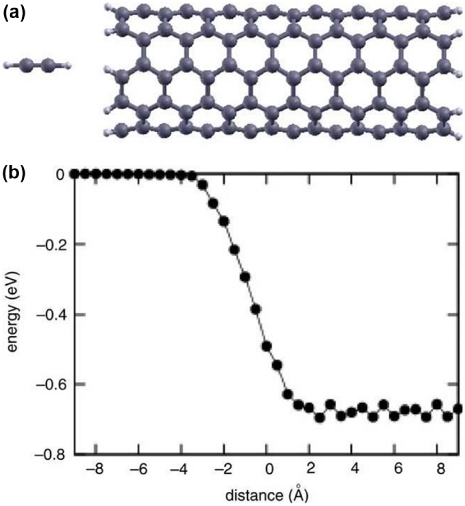
Figure 10. Energetics of acetylene insertion into a CNT. (a) Atomic model of a C2H2 molecule entering a (5,5) nanotube. (b) Energy profile during the insertion of C2H2 along the tube axis. Figure reprinted from [74].
Download figure:
Standard image High-resolution imageNotice that in another theoretical work [75], the characterization of carbon chain molecules encapsulated in an SWCNT has been achieved using the DMOL program [76]. They show that polyynes and their dehydrogenated forms can be spontaneously encapsulated in an open-ended SWCNT. No polymerization is determined in this work, but the encapsulation reaction is also found to be exothermic, confirming the results found in the above cited work and showing that hydrogenocarbonated molecules encapsulation is an energetically favourable process.
As an extrapolation, we can remark that a specific graphitic molecule can be constituted by another nanotube inserted in the host nanotube. This system has been studied theoretically as a reference system using the LCAO-S2 + vdW by calculating the inter-wall energy of a DWCNT like (4,4)@(10,10). The resulting cohesion energy is 0.92 eV Å−1, which is totally comparable with the one obtained with the fullerene encapsulation in the same case [50].
To close this section, we will finish by presenting theoretical calculations on polythiophene encapsulation in CNTs. In a first study [77], terthiophene molecules are inserted in zig-zag CNTs with diameters ranging from 8 to 13 Å. Calculations have been performed using the SIESTA code without inclusion of vdW forces [78]. Here also the encapsulation process is shown to be exothermic as represented in figure 11 for a cleaned or hydrogenated opened SWCNT.
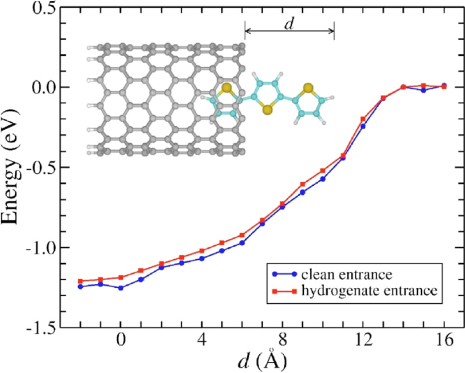
Figure 11. Energy profile during the axial insertion of the terthiophene molecule into the (13,0) CNT by both clean and hydrogenate entrances. The inset shows the terthiophene molecule entering inside the (13,0) CNT by the clean entrance. Figure reprinted from [77].
Download figure:
Standard image High-resolution imageSimilar energy profiles are observed in both cases, indicating that electrostatic forces induced by the dangling bonds at the SWCNT termination are not determinant for the insertion process. This means again that vdW forces dominate the molecular encapsulation process in CNTs. It is observed then that the incorporation reaction is exothermic by 1.2 eV. Thus, it is energetically more favourable that the molecule enters into an uncapped nanotube than remains outside, exhibiting a barrierless insertion process. Since the CNT is slightly charged due to a curvature effect, the encapsulation of the terthiophene is driven by nanocapillarity forces, induced by the interaction between both electronic densities of the nanotube and the molecule. Moreover, band-structure calculations show that the electronic structure of the molecule is preserved through encapsulation.
The second work on polythiophene encapsulation in CNTs present a general scheme for predicting the electronic structure of vdW bound systems, using this example of encapsulation [79]. They have therefore proceeded to DFT calculations, using the QUANTUM-ESPRESSO package [80] and considering vdW forces through the use of the vdW-DF method in a post-GGA manner. Several SWCNTs of (n, 0) type have been considered, with n ranging from 10 to 22. Here as well, the exothermic character of the process is exhibited, and an electronic structure study is performed to show the negligible charge transfer between the tube and the molecule due to vdW interactions.
In the next sections, we will precisely consider those electronic aspects in the encapsulation process. We have seen many examples of structures and configurations, we will now consider some possible applications as consequences of these structural aspects, as magnetic, charge transfer and electronic transport, and optical properties of encapsulated molecules in CNTs.
4. Encapsulated molecules and magnetic properties
Among the numerous applications of molecules inserted in CNTs, lie also the magnetic aspects in these systems. In this section we will only say some words about properties of magnetic molecules encapsulated in SWCNT through specific examples.
Single-molecule magnets (SMM) constitute a wide range of new materials for applications in future magneto-electronics or data storage devices. An important issue however, is to connect these molecules, since they are quite often decomposed by deposition on a conductive surface. CNTs offer an excellent solution to connect these molecules, since vdW interactions are weak enough to not destroy molecular functionalities. In a work presented by Giménez-López et al [10], Mn12Ac encapsulation in graphitized multi-walled CNT (GMWNT) is considered, as represented in figure 12.

Figure 12. Chemical structure of Mn12Ac SMM and schematic representation of its encapsulation in a CNT as studied in [10]. (a) Ball and stick structure of [Mn12O12(O2CCH3)16(H2O)4] (Mn12Ac), with its dimensions indicated by double-headed arrows. Carbon, hydrogen, manganese and oxygen atoms are shown as grey, light-blue, purple and red circles, respectively. (b) Schematic representation of the innermost layer of a GMWNT hosting Mn12Ac molecules.
Download figure:
Standard image High-resolution imageMn12Ac is by far the most-studied SMM and exhibits a blocking temperature as high as 4 K, which is much higher than that of the vast majority of other SMM. After encapsulation, the systems have been observed by means of HRTEM showing no degradation of the inserted molecules. Under an electron beam, molecules have been observed to move within the tube, which is another proof that molecules are held inside the tube by vdW forces. Magnetic properties have also been extensively studied. Therefore, a hysteresis cycle has been recorded at 1.8 K, with sizeable coercivity of 0.2 T. In the same manner, a significant negative magnetoresistance is also observed, in sharp contrast with the isolated nanotube. Consequently, this study describes a methodology for integration of SMM into the internal cavity of CNTs. The non-covalent interactions, which are responsible for efficient transport and encapsulation of the guest molecules into nanotubes, allow the molecules to stay mobile within the nanotube and align along the applied magnetic field. Although the encapsulated Mn12Ac molecules retain their SMM properties, the host nanotubes provide an alternative pathway for relaxation of SMM magnetization. In return, the magnetic guest molecules influence the electrical resistance of the host nanotubes. Therefore, the new class of hybrid structures SMM@GMWNT represents a synergistic material combining the functional properties of nanotubes and magnetic molecules.
This study is following a previous work on the encapsulation of cobalt phthalocyanine (CoPc) molecules in multi-walled CNTs (MWCNTs) [11]. In this experiment, CoPc@MWCNT have been characterized by HRTEM and core-level photoemission spectroscopy. The results show an hybridization of the Co 2p orbitals with the surrounding nanotube, opening new possibilities for magnetic properties of encapsulated molecules in CNT.
Finally, we can mention a theoretical investigation on doped CNTs by MCp2 (M = Fe, Co, Ni) molecular encapsulation [81]. In this work, confined metallocenes structural, energetics and electronic properties have been studied by the mean of DFT. Namely, the calculations have been performed using the DMOL package, without inclusion of vdW interactions. However, as we said earlier, this approach can yield the good tendency for geometry optimization. Several zig-zag nanotubes have been considered here. This work demonstrates the non-covalent aspects of the molecule–tube interaction, as well as new possibilities for electronic and magnetic properties of magnetic molecules encapsulated in CNTs.
5. Encapsulated molecules and electrical properties: charge transfer and electronic transport
In this section, we will focus particularly on the electrical properties of encapsulated molecules in CNTs. Our purpose is to give some examples of electric modification in the nanotube due to the molecule, that is to say charge transfer and electronic transport properties. Generally speaking, since those equilibrium are ruled by vdW, the molecular electronic structure is only slightly or not at all affected by the interaction. This is not always the case, as we will see in the optical section. However, even without important modifications, charge transfer occurs between the tube and the molecule, which can affect the electric conductance of the tube. The charge transfer properties represent then an important aspect for applications in molecular electronics [82, 83]. For example, it has been observed that a C60 in a peapod structure presents an excess of electronic charge, resulting in a negative net charge [84, 85].
This feature has been demonstrated also theoretically [86] where peapods based on metallic and semiconducting nanotubes have been simulated by the mean of molecular dynamics and extended Hückel (EH) methods. The electrical transport properties of those peapods were computed using a Green's function approach within the Landauer–Büttiker formalism. The Hamiltonian and overlap matrices used in this formalism were also determined using the EH model. Transport calculations have been performed connecting the two extremities of the peapods with gold electrodes. As a result, a weak charge transfer from the nanotube to the fullerenes is observed, which increases as the tube diameter decreases, until the thermodynamic stability limit. Regarding the transport properties, the smaller diameter peapods are also more influenced, but in general the changes in the conductance remain quite weak, due probably to the fact that we are considering interactions between the same species (carbon).
Similarly, we have studied with the LCAO-S2 + vdW formalism the encapsulation and structural properties of an anthracene molecule in an SWCNT [87]. Standard DFT calculations have also been performed by means of the SIESTA code. The transport properties of the CNT have been determined using the non-equilibrium Green's-function method (NEGF) as implemented in the SMEAGOL code [88]. As a first step, the equilibrium configuration of the anthracene molecule in the CNT has been determined. It shows a rotation angle of the molecule with respect to the tube axis of about 40° (figure 13). This is due to the particular rectangular shape of the molecule, and the balance between repulsive forces associated to the electronic densities overlaps and the attractive forces related to the pure vdW interaction. Notice that no clear minimum is found when vdW forces are not taken into account.
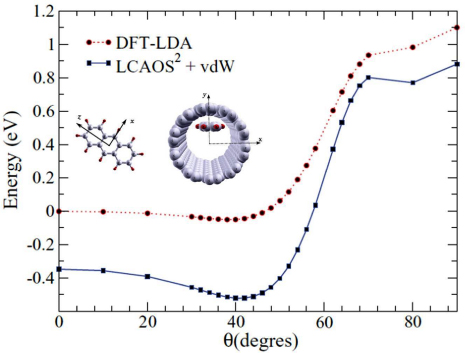
Figure 13. Binding energy (eV) of the anthracene molecule encapsulated inside the SWCNT as a function of the orientation angle in the (x, z) plane. The red curve with circles represents the LDA result, and the blue curve with squares represents that obtained by means of the LCAO-S2 + vdW method. In inset, structure of the isolated anthracene molecule and its encapsulation inside the (10,10) SWCNT, as used in the calculations from [87].
Download figure:
Standard image High-resolution imageConsidering this behaviour, results of conductance calculations at the equilibrium position and also in the case of a strong repulsive configuration, around 80°, are presented in figure 14. The objective here is to correlate the difference in the strengths of interaction with the calculated electric conductances.

Figure 14. Comparison of transmission coefficients calculated in [87] for the isolated SWCNT and for the combined system SWCNT + anthracene in the two different configurations (θ = 40° and 80°).
Download figure:
Standard image High-resolution imageAs is well known, the isolated pristine SWCNT presents a transmission of 2G0, with G0 = 2e2/h being the quantum of conductance. In figure 14 we can observe the reduction of conductance from 2G0 to 1G0 at the new energy levels induced by the coupling between the molecule and the nanotube. These localized states are due to the interaction of the HOMO of the molecule with the electronic structure of the nanotube and to the resulting charge transfer from the nanotube to the anthracene molecule. They can scatter an incoming conduction electron and thus reduce the conductance by one quantum conductance. This effect becomes greater when the molecule approaches the nanotube wall. In particular it clearly appears when the molecule varies from 40° to 80°. This reduction in the transmission is pushed towards higher energies for θ = 80° due to the strong interaction of the molecule with the nanotube.
A similar system has been studied also by standard DFT calculations using the VASP code. In this work, the authors have considered the encapsulation of F4-TCNQ molecule in several types of SWCNTs, either zig-zag or armchairs [89]. This molecule is interesting because it is kind of rectangular as the anthracene previously considered, but it has two different terminal groups on the large and short sides. This adds an extra contribution to the equilibrium, due to the different polarizabilities of the groups. Several configurations have been considered, but the molecule results to lie always in a flat position, parallel to the tube axis. Maybe here there is no rotation angle due to the different chemical groups which balance the geometry of the molecule. Regarding the electronic structure, this molecule appears to introduce impurity bands near the Fermi level of the nanotube, which are attributed to the π bands of the molecule. As a consequence, a small charge transfer occurs between the SWCNT and the molecule, resulting in a small p-doping of the nanotube.
Finally, we will close this section with a really interesting work from Yanagi et al [90] where they present charge manipulation of β-carotene (Car) encapsulated in a CNT, by the mean of electrochemical doping technique. By applying different electric fields on the hybrid tube–molecule system, they have extracted one electron from the Car molecule and observed the influence on the electronic structure of the system. Supporting DFT calculations show that the HOMO band of the Car was located below the valence band of the SWCNT when the Fermi level was shifted. The evolution of the electronic structure with the applied electric field is represented in figure 15.
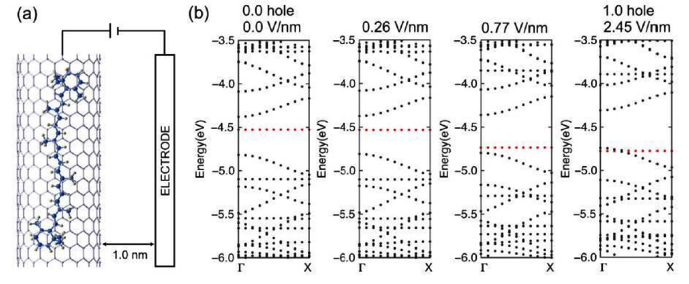
Figure 15. (a) Schematic representation of the calculations. (b) Electronic structures of Car@(17,0) under various electric fields. The red dots correspond to the HOMO band of Car, and the black dots correspond to (17,0). At 2.45 V nm−1, one electron was extracted from Car. Reprinted with permission from [90]. Copyright 2013 American Physical Society.
Download figure:
Standard image High-resolution imageWe can observe the evolution of the band structure under application of an electric field, until one electron is extracted. The shift of the Fermi level appears clearly as well as the moving of the HOMO band. This study elucidates the process of electron extraction for encapsulated molecules in CNTs, opening the way for charge manipulation in molecular devices.
6. Encapsulated molecules and optical properties
Now we will turn to the description of some optical properties of encapsulated molecules in SWCNTs which constitutes an important aspect of these systems. In particular, CNTs are really well characterized by Raman spectroscopy. This represents a fantastic tool to determine the fundamental properties of these nanomaterials. For example, it includes the determination of their diameter and chirality, an assessment of structural order/disorder and also whether their electronic behaviour is metallic or semiconductor in nature. Therefore, the encapsulation of a molecule which can affect the electronic properties of the nanotube can obviously be observed by Raman spectroscopy.
For example, the Raman resonance of polyyne molecules encapsulated in CNTs has been studied using many different laser lines in the visible range [91]. The resonance spectra of the C10H2 and C12H2 molecules encapsulated inside SWCNTs has been measured. A strong enhancement of the stretching modes of the linear chains is observed around 2.1 eV. This value is much lower than the expected value around 5 eV in gas phase. This is interpreted as a dipole-forbidden transition due to a symmetry breaking when the molecules are encapsulated. This result shows the strong influence of the encapsulation on optical properties of molecules.
Another example is the π-stacking of coronene molecules encapsulated in CNTs [92]. In this theoretical work, based on experimental results from Okazaki et al [93] we address the problem of absorption spectra of encapsulated coronenes C24H12@SWCNT(19,0). First we analyse the structure of the coronene column in the nanotube using the LCAO-S2+ vdW formalism. We have demonstrated in particular that the equilibrium of the system is due to a 30° rotation angle between one coronene and its nearest coronene neighbour. This rotation which has not been seen experimentally is absolutely fundamental for the stability of the system and is ruled by vdW interactions. We also find an intermolecular distance of 4 Å, and a 35° tilt angle, as shown in figure 16.
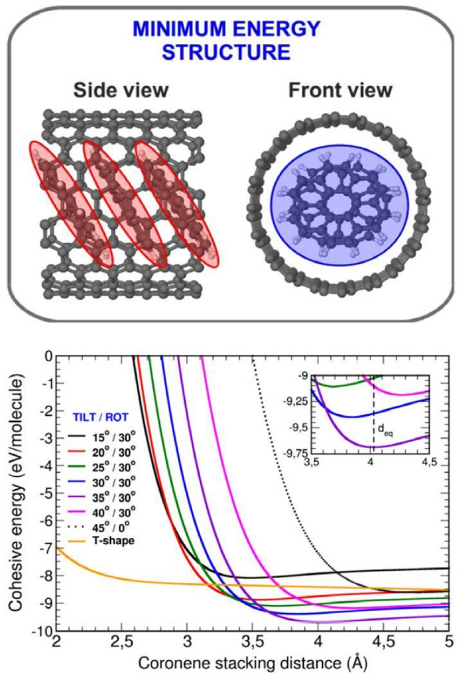
Figure 16. (Bottom panel) Cohesive energy per coronene molecule (inserted between two neighbouring coronenes) as a function of the intermolecular stacking distance of an infinite chain of stacked C24H12 coronenes encapsulated inside the (19,0) SWCNT. Cohesive energy is depicted for several tilt angles, considering a rotation angle of 30° between the coronenes. Most stable structure (sketched in top panel) is obtained for a 35° tilt angle, at an intermolecular stacking distance of around 4 Å. Total energy minimum region has been scale-enlarged as an inset in middle panel for a better visualization. Figure reprinted from [92].
Download figure:
Standard image High-resolution imageIn a second step, we have analysed the electronic structure of the system as well as the excitation spectrum through the use of advanced many body techniques like the quasi-particle Green function screened interaction approximation (GW) [94], combined to the Bethe–Salpeter equation (BSE) [95]. The results of these calculations are presented in figure 17.
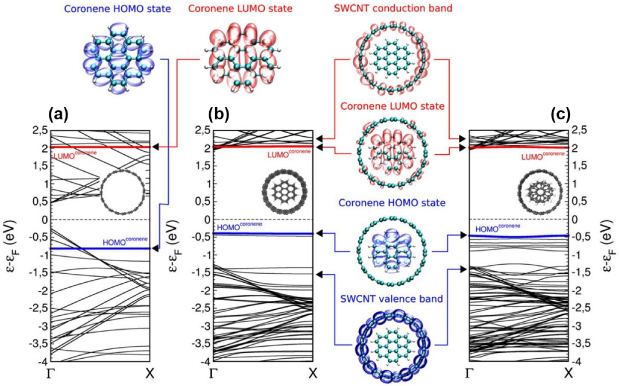
Figure 17. Quasi-particle GW-corrected DFT band-structure diagrams along the most relevant symmetry line from Γ → X (along nanotube axis) (with respect to the Fermi level energy) for: (a) Isolated (19,0) SWCNT. HOMO and LUMO levels of the self-assembled stacked C24H12 coronenes (dilute phase) are also shown superimposed onto the nanotube band diagram; (b) (19,0) SWCNT@C24H12 interacting system (θtilt = 30°/θrot = 0°); and (c) (19,0) SWCNT@C24H12 interacting system (θtilt = 35°/θrot = 30°). Additionally, 3D orbital isodensities, all of them with a value of 2 × 10−5 e− Å−3, are shown for the most representative states of the systems. HOMO and LUMO coronene states have been visually emphasized within all the band diagrams (solid blue and red lines, respectively). Figure reprinted from [92].
Download figure:
Standard image High-resolution imageIn a really interesting manner, we can observe a global 'dilatation' of the nanotubes bands, meaning that unoccupied states move upwards and occupied states move downwards, meanwhile the HOMO–LUMO gap is being reduced by the interaction with the nanotube. Note however that there is no influence of intermolecular interactions between coronenes, since in the determined configuration or with the corresponding rotation angle, there is no difference in the band structure of the system. This small closing of the gap is expressed by a red-shift in the excitation spectra, from 2.97 eV in the coronene gas phase to 2.49 eV for the encapsulated phase. This result is in very good agreement with experimental results [93] since coronenes in gas phase emit light in the blue range, whereas they emit in the green range when encapsulated.
As a last example, we will close this section by very recent study of optical properties of GNRs encapsulated in an SWCNT [96]. In this work, the authors report a photoluminescence study of GNRs encapsulated in SWCNTs. They demonstrate that GNR@SWCNT can be characterized by photoluminescence mapping and observe new spectral features in the visible range. Those peaks have resonant character and their positions depend on the GNR geometrical structure. As a consequence, this work opens a wide range of applications for encapsulated GNR in SWCNTs in photonics or optoelectronics.
7. Encapsulated molecules and biomaterials
In this last section, we will present some examples of applications for encapsulated molecules in biomaterials. Indeed, the nanotubes can be used for drug delivery in the human body or to study isolated biological molecules. Those biological molecules can even be used as building blocks for new electronic devices, like standard molecules, but also for DNA as electronic DNA sequencing or gene delivery systems for example. Another application is the transport of DNA through biological membranes, by the mean of the nanotube.
As an illustration, we can mention a work on total energy electronic structures calculations based on DFT. The calculations have been performed on a single strand DNA (ss-DNA) encapsulated in an SWCNT [97]. They have considered four different types of ss-DNA, as represented in figure 18, and determined their properties after encapsulation in SWCNTs.
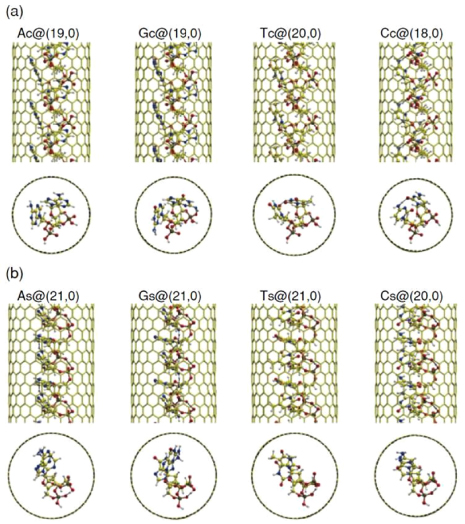
Figure 18. Optimized geometries of ss-DNA encapsulated in their corresponding energetically optimized nanotubes for the (a) coating and (b) stacking ss-DNA configurations. A complete circle is shown by dashed lines, superimposed on nanotubes in the top view panels. Pairs of atoms satisfying the H-bond criteria, i.e. separated by 3.2 Å, with a bonding angle of 120°, are connected by dotted lines. Here and in the following figures, the colour code for the atoms is yellow for C, white for H, red for O, blue for N, and tan for P, respectively. Reprinted with permission from [97]. Copyright 2011 American Physical Society.
Download figure:
Standard image High-resolution imageAs a result, it is shown that the encapsulation process is exothermic, allowing the filling of nanotubes with DNA strands. Regarding the electronic structures, even though it remains roughly the same, they can observe some shifts in the HOMO level or in the nanotube gap. A charge transfer is associated to these shifts as described earlier for other types of molecules. This opens the way to DNA@SWCNT based nanoelectronics.
In the same manner, some works have been dedicated to the filling of nanotubes with water molecules, as a nanorecipient. The study of confining water in CNTs presents two major interests: first, due to the confinement, water molecules are expected to get different physical properties such as important changes in the phase diagram. Second, nanotube dimensions are comparable to the ones of ion channels in natural biological environment. Therefore, water in nanotube represents a fantastic system for fundamental description of water behaviour in complex biological system. In this work, water molecules confined in single-walled (6,6) CNTs [98] have been considered from first-principles study. Namely, the DMol program has been used, as an all-electron method without inclusion of vdW energy. For one water molecule encapsulation, the equilibrium geometry is found to be perpendicular to the tube axis. With more water molecules, the hydrogen bonds between molecules form a one-dimensional chain confined in the tube, with 0.1 to 0.28 eV coupling energy per molecule (depending on the functional used for the calculations). Moreover, the electron transfer from hydrogen to oxygen is slightly enhanced with respect to the isolated molecule. This study shows also that even though the water–tube interaction is mainly vdW, the confinement in the tube also affects electronic and vibrational properties of the water molecules.
As a last interesting work to mention, we present now the encapsulation of a lamivudine molecule in an SWCNT [99]. Lamivudine belongs to a class of drugs named nucleoside analogues which has been approved for the treatment of HIV. The aim of this study is to characterize the behaviour of these molecules inserted in nanotubes for applications in drug delivery using the nanotube as a transporter. In particular, they have considered the inclusion of this molecule in several kinds of nanotubes as represented in figure 19.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 19. Representation of the calculated bonding distances between two closest atoms of the interacting entities for the (left panel) lamivudine@CNT(10,10) and (right panel) lamivudine@CNT(14,0) complexes. Figure reprinted from [99].
Download figure:
Standard image High-resolution image{kind=link}
Calculations have been performed using the SIESTA code including the vdW-DF functional for vdW energy calculations. Among all the considered metallic and semiconducting nanotubes, the (10.10) and (14,0) form the most thermodynamically stable complexes. A slightly stronger interaction is found for the metallic tube. Finally, the electronic structure of the complexes has been analysed, revealing no considerable orbital hybridization between the molecule and the tube. As a consequence, the nanotube constitutes a perfect host for the Lamivudine molecule, with potential applications for drug delivery in the human body.
8. Summary
In this review we have presented a general overview of encapsulated molecules in CNTs. We place special emphasis on weak and vdW interactions, since these interactions play a fundamental role in the molecular encapsulation. Therefore we have described the important methods based on DFT to treat these kind of interactions, and we have presented some applications of these hybrid systems. We have considered the structural aspects of these systems, such as peapods or molecular chains in nanotubes, and stressed the non-covalent character of the molecule–tube coupling. An interesting application is also the polymerization of graphitic-like molecules to form graphene nanoribbons. Then we have seen several examples of insertion of molecular magnets in nanotubes in order to conceive magnetic devices for data storage. Transport properties and charge transfer have also been discussed with various examples. We have seen how transport properties can be affected by the molecular insertion, as well as how molecules electronic structure can be slightly modified. We have also seen the influence of encapsulation on optical properties of the complex system, in particular for Raman spectroscopy or red-shift in the photoexcitation spectrum. Finally, we have considered applications in biomaterials, with the confinement of DNA molecule in nanotube, or the study of molecules for drug delivery.
Through these few examples, we can notice that molecular encapsulation offers a wide range of applications for future materials and technological applications. This can be seen as a particular case of supramolecular assembly where the number of combinations between molecules and nanotubes is incredibly huge. Now the next perspective will be the combination of these new hybrid systems, such as connecting several nanotubes with different encapsulated molecules, to design new molecular electronic devices or new biomembranes.